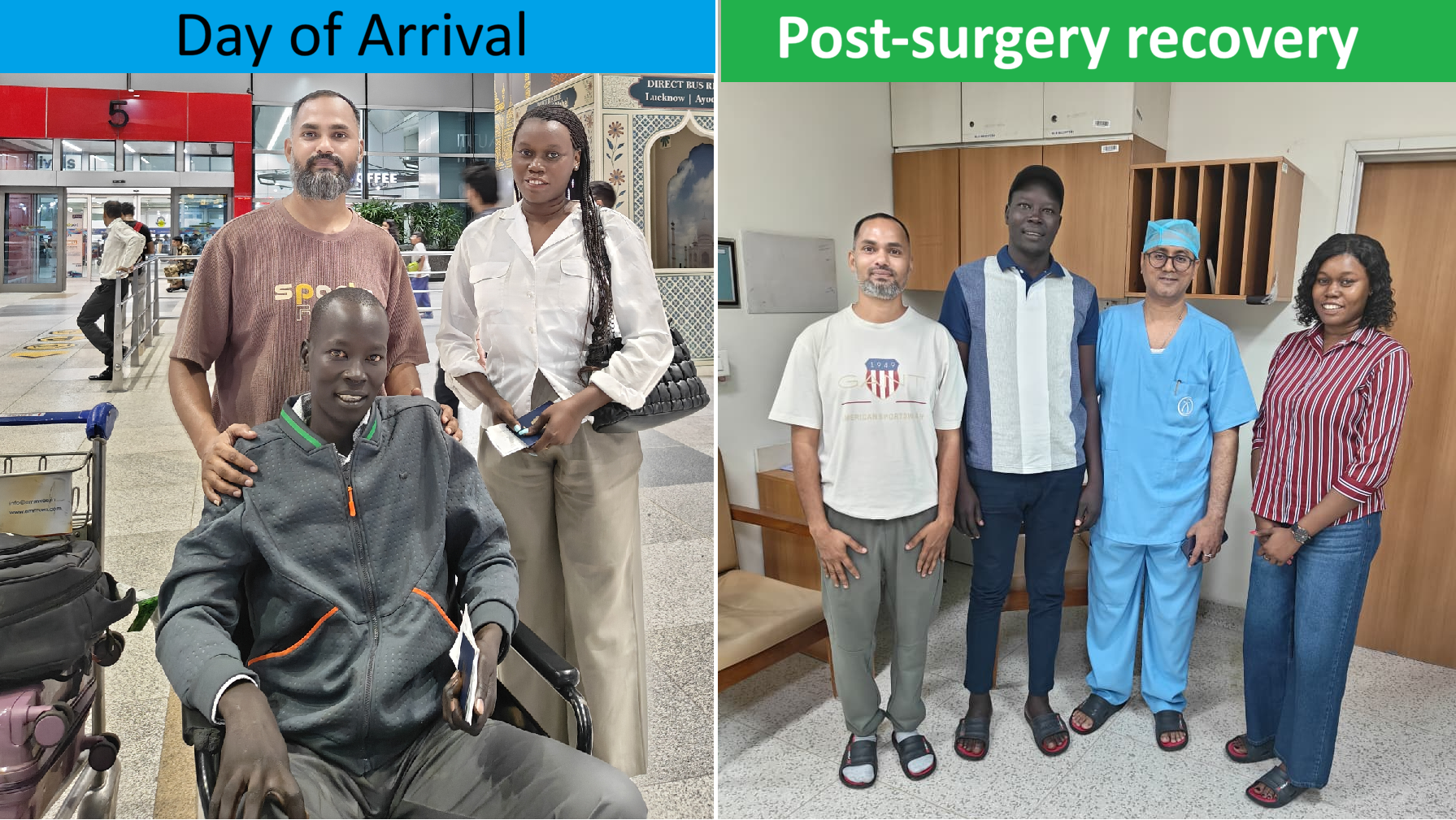
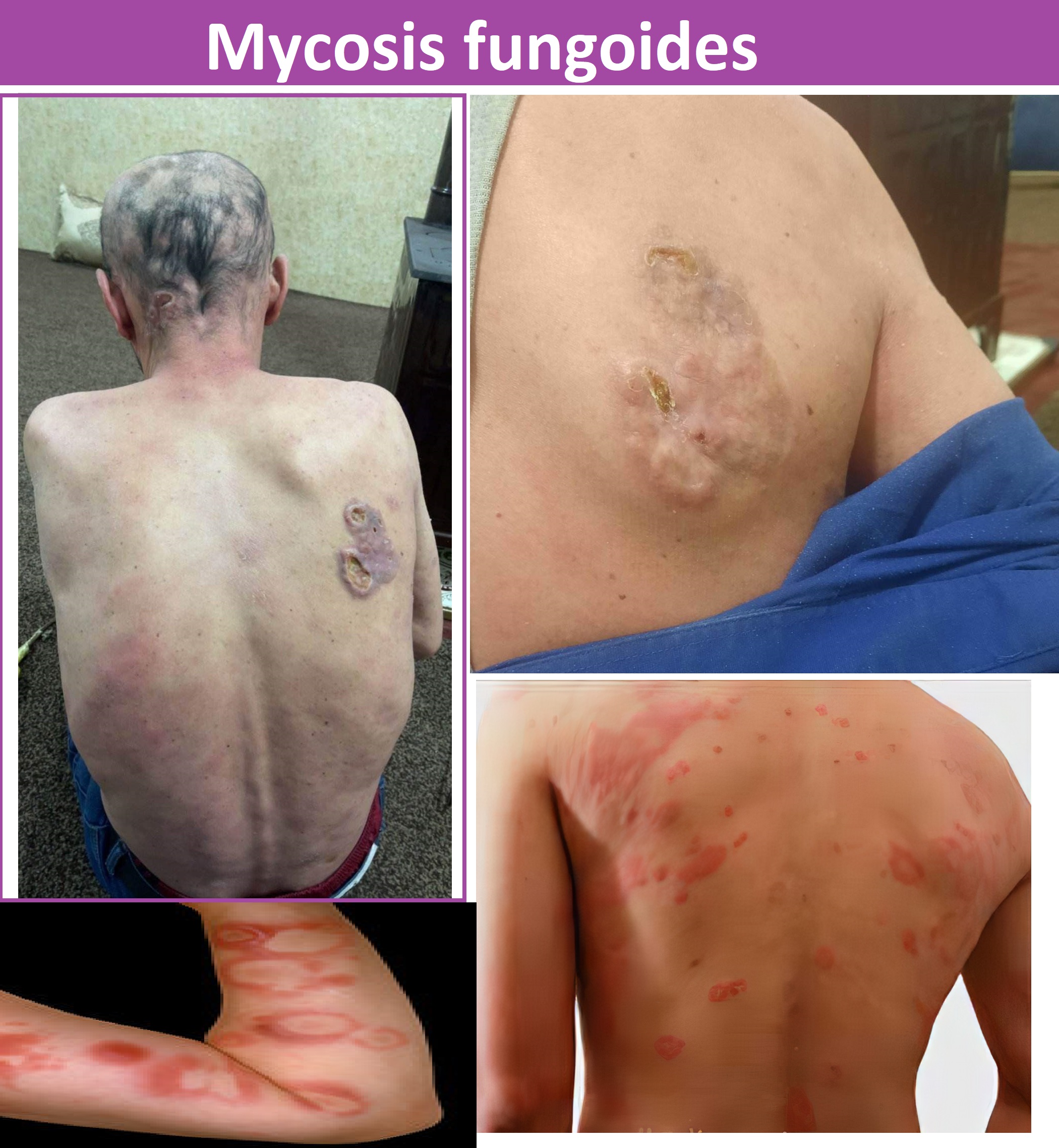
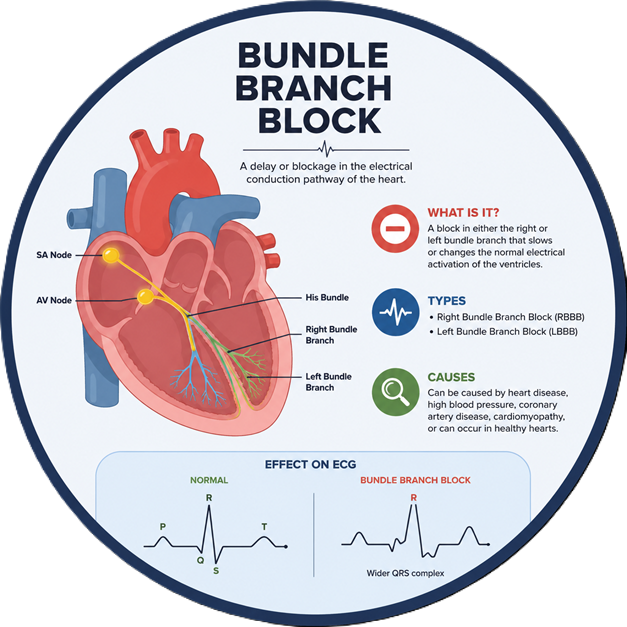
Gender
Male
Female
Others
+0
+1
+1242
+1246
+1264
+1268
+1284
+1340
+1345
+1441
+1473
+1649
+1664
+1670
+1671
+1684
+1758
+1767
+1784
+1787
+1809
+1868
+1869
+1876
+20
+212
+213
+216
+218
+220
+221
+222
+223
+224
+225
+226
+227
+228
+229
+230
+231
+232
+233
+234
+235
+236
+237
+238
+239
+240
+241
+242
+244
+245
+246
+248
+249
+250
+251
+252
+253
+254
+255
+256
+257
+258
+260
+261
+262
+263
+264
+265
+266
+267
+268
+269
+27
+290
+291
+297
+298
+299
+30
+31
+32
+33
+34
+350
+351
+352
+353
+354
+355
+356
+357
+358
+359
+36
+370
+371
+372
+373
+374
+375
+376
+377
+378
+380
+381
+385
+386
+387
+389
+39
+40
+41
+420
+421
+423
+43
+44
+45
+46
+47
+48
+49
+500
+501
+502
+503
+504
+505
+506
+507
+508
+509
+51
+52
+53
+54
+55
+56
+57
+58
+590
+591
+592
+593
+594
+595
+596
+597
+598
+599
+60
+61
+62
+63
+64
+65
+66
+670
+672
+673
+674
+675
+676
+677
+678
+679
+680
+681
+682
+683
+684
+686
+687
+688
+689
+690
+691
+692
+7
+70
+7370
+81
+82
+84
+850
+852
+853
+855
+856
+86
+880
+886
+90
+91
+92
+93
+94
+95
+960
+961
+962
+963
+964
+965
+966
+967
+968
+970
+971
+972
+973
+974
+975
+976
+977
+98
+992
+994
+995
+996
+998
---Select Country---
Afghanistan
Albania
Algeria
American Samoa
Andorra
Angola
Anguilla
Antarctica
Antigua And Barbuda
Argentina
Armenia
Aruba
Australia
Austria
Azerbaijan
Bahamas The
Bahrain
Bangladesh
Barbados
Belarus
Belgium
Belize
Benin
Bermuda
Bhutan
Bolivia
Bosnia and Herzegovina
Botswana
Bouvet Island
Brazil
British Indian Ocean Territory
Brunei
Bulgaria
Burkina Faso
Burundi
Cambodia
Cameroon
Canada
Cape Verde
Cayman Islands
Central African Republic
Chad
Chile
China
Christmas Island
Cocos (Keeling) Islands
Colombia
Comoros
Republic Of The Congo
Democratic Republic Of The Congo
Cook Islands
Costa Rica
Cote D'Ivoire (Ivory Coast)
Croatia (Hrvatska)
Cuba
Cyprus
Czech Republic
Denmark
Djibouti
Dominica
Dominican Republic
East Timor
Ecuador
Egypt
El Salvador
Equatorial Guinea
Eritrea
Estonia
Ethiopia
External Territories of Australia
Falkland Islands
Faroe Islands
Fiji Islands
Finland
France
French Guiana
French Polynesia
French Southern Territories
Gabon
Gambia The
Georgia
Germany
Ghana
Gibraltar
Greece
Greenland
Grenada
Guadeloupe
Guam
Guatemala
Guernsey and Alderney
Guinea
Guinea-Bissau
Guyana
Haiti
Heard and McDonald Islands
Honduras
Hong Kong S.A.R.
Hungary
Iceland
India
Indonesia
Iran
Iraq
Ireland
Israel
Italy
Jamaica
Japan
Jersey
Jordan
Kazakhstan
Kenya
Kiribati
Korea North
Korea South
Kuwait
Kyrgyzstan
Laos
Latvia
Lebanon
Lesotho
Liberia
Libya
Liechtenstein
Lithuania
Luxembourg
Macau S.A.R.
Macedonia
Madagascar
Malawi
Malaysia
Maldives
Mali
Malta
Man (Isle of)
Marshall Islands
Martinique
Mauritania
Mauritius
Mayotte
Mexico
Micronesia
Moldova
Monaco
Mongolia
Montserrat
Morocco
Mozambique
Myanmar
Namibia
Nauru
Nepal
Netherlands Antilles
Netherlands The
New Caledonia
New Zealand
Nicaragua
Niger
Nigeria
Niue
Norfolk Island
Northern Mariana Islands
Norway
Oman
Pakistan
Palau
Palestinian Territory Occupied
Panama
Papua new Guinea
Paraguay
Peru
Philippines
Pitcairn Island
Poland
Portugal
Puerto Rico
Qatar
Reunion
Romania
Russia
Rwanda
Saint Helena
Saint Kitts And Nevis
Saint Lucia
Saint Pierre and Miquelon
Saint Vincent And The Grenadines
Samoa
San Marino
Sao Tome and Principe
Saudi Arabia
Senegal
Serbia
Seychelles
Sierra Leone
Singapore
Slovakia
Slovenia
Smaller Territories of the UK
Solomon Islands
Somalia
South Africa
South Georgia
South Sudan
Spain
Sri Lanka
Sudan
Suriname
Svalbard And Jan Mayen Islands
Swaziland
Sweden
Switzerland
Syria
Taiwan
Tajikistan
Tanzania
Thailand
Togo
Tokelau
Tonga
Trinidad And Tobago
Tunisia
Turkey
Turkmenistan
Turks And Caicos Islands
Tuvalu
Uganda
Ukraine
United Arab Emirates
United Kingdom
United States
United States Minor Outlying Islands
Uruguay
Uzbekistan
Vanuatu
Vatican City State (Holy See)
Venezuela
Vietnam
Virgin Islands (British)
Virgin Islands (US)
Wallis And Futuna Islands
Western Sahara
Yemen
Yugoslavia
Zambia
Zimbabwe
---Opinion from Doctor---
Dr. Arvind Sabharwal
Dr. Dhananjay Kumar Jhamb [MBBS, MD - Medicine, DM - Cardiology]
Dr. Prashant Jain
Dr. Rahul Naithani [DM - Clinical Haematology, MD - Pediatrics, MBBS]
Dr. Rekha Mittal [MBBS, MD - Pediatrics, MBBS]
Dr. Saurabh Kapoor [MBBS, FRCS , MS (Gold Medallist), MRCS, DNB (Orthopaedics)
Dr. Suman Lata Nayak
Aakriti Aggarwal [MSc Medical Genetics, University of Glasgow UK; Certified Board of Genetic Counseling]
ACHARYA DEEPAK SINGH
Dr Anand Shukla
Dr Ashok Seth
Dr Gaurav Gangwani [Interventional Radiologist]
Dr Gaurav Gupta
Dr Gautam Niloba Naik [MBBS, MD - General Medicine, DM - Cardiology]
Dr Harsh Bhargava [MBBS, MS - Orthopaedics, AO Fellowship - Traumatology]
Dr Kuldeep Sharma [MD - Radiotherapy, MBBS]
Dr Muthu Jothi [MBBS, MS - General Surgery, MCh - Cardiothoracic Surgery, Pediatric Cardiac Surgeon]
Dr Naveen Verma
Dr Nidhi Rajotia Goel [MBBS, MD - Obstetrics and Gynecology, DNB - Obstetrics and Gynecology]
Dr Rajesh Goel
Dr Rajesh Sharma
Dr Randeep Wadhawan
Dr Rita Bakshi [MBBS, MD - Obstetrics & Gynaecology,VF Specialist]
Dr Rupesh kaushik [MBBS, MD - General Medicine, DM - Cardiology]
Dr S Towheed
Dr Shailendra Lalwani [MBBS, MS - General Surgery , DNB - Surgical Gastroenterology]
Dr Shiveta Razdan
Dr Sumit Mrig [ENT, COCHLEAR IMPLANT & SLEEP APNEA SURGERY]
Dr Vivek Verma
Dr(Prof) Amit kumar Sharma [MBBS, MS ENT | ENT, Head and Neck Surgeon]
Dr. (Air Cmde) Krishan Kumar Yadav [MBBS, MS - General Surgery, MCh - Neurosurgery, FAIS]
Dr. (Brig) Anil Kumar Dhar [MBBS, MD - Internal Medicine (KG's Medical College, Lucknow, 1983), Post-Doctoral Fellowship in Medical Oncology (Tata Memorial Hospital, Mumbai, 1994), VSM]
Dr. (Brig) Sameer Kumar [MBBS, MS - General Surgery, MCh - Cardiothoracic Surgery, Armed Forces Medical Services]
Dr. (Brig.) Anil Khetarpal
Dr. (Brig.) B. K. Singh
Dr. (Col) Balbir Kalra [MBBS, MD - General Medicine, DM - Cardiology]
Dr. (Col) Joy Dev Mukherji [MBBS, MD - General Medicine, DM - Neurology]
Dr. (Col) Manjinder Sandhu
Dr. (Col) Preema Sinha
Dr. (Col) R. Ranga Rao [MBBS, MD - Internal Medicine, DM - Medical Oncology]
Dr. (Col.)Arun Kumar
Dr. (Lt Col) Aditya Pradhan
Dr. (Lt Gen) CS Narayanan
Dr. (Lt. Gen.) Prem Prakash Varma
Dr. (Maj.) Mukesh Garg
Dr. (Prof) Daljit Singh [MBBS, MS – General Surgery, MCh – Neurosurgery]
Dr. (Prof) Sampath Chandra Prasad Rao [MBBS, MS - ENT, DNB - Otolaryngology]
Dr. (Prof) Sumit Sinha [MBBS, MS - General Surgery , DNB - General Surgery]
Dr. (Prof) Vinit Banga [MBBS, MD - General Medicine, DM - Neurology]
Dr. (Prof.) A. K. Banerji
Dr. (Prof.) Arvind Kumar [MBBS, MS - General Surgery]
Dr. (Prof.) K. N. Srivastava
Dr. (Prof.) Sanjay Kumar Agarwal [MBBS, MD, DM - Nephrology (AIIMS), 40+ years at AIIMS New Delhi]
Dr. (Prof.) Shashank Sharad Kale [MBBS, MS-General Surgery, MCh. - Neurosurgery]
Dr. A K Bardhan
Dr. A K Gadpayle [MBBS, MD - Medicine ]
Dr. A Sharath Reddy
Dr. A Sivagnanam [MBBS, MD, DNB - Paediatrics]
Dr. A. Anbanandan
Dr. A. Mohamed Ibrahim
Dr. A. S. Bhagwat [BDS, MDS]
Dr. A. Suresh
Dr. Aadarsh Raghavan
Dr. Aakanksha Chawla Jain [MD (Pulmonary Medicine), IDCCM, IFCCM (Indian Fellowship in Critical Care Medicine)]
Dr. Aanchal Agarwal [MBBS, DGO, DNB, FNB-Reproductive Medicine , Reproductive Endocrinology, Fertility & IVF Specialist ]
Dr. Aarti Nangia
Dr. Aarushi Garg [MBBS, MD - Radiology, DNB - Radiology]
Dr. Aashish Chaudhry
Dr. Aayush Chawla
Dr. Aayush Gupta [MBBS, MD - Internal Medicine]
Dr. Abdul S. Ansari
Dr. Abhai Verma
Dr. Abhay G Uppe [MBBS, MD - Chest Medicine, TDD]
Dr. Abhay Huprikar [MD Medicine, DM Nephrology]
Dr. Abhay Jain
Dr. Abhay Kapoor
Dr. Abhay Kulkarni [MBBS, MS (Orthopaedics), MRCS (Edinburgh, UK), Fellowship Shoulder Surgery & Sports Medicine]
Dr. Abhay Kulkarni [MS Orthopaedics]
Dr. Abhay Kumar
Dr. Abhay Sadre [MD, DM Nephrology]
Dr. Abhay Somani [MBBS, DNB - General Medicine, DNB - Cardiology]
Dr. Abheek Kar
Dr. Abhideep Chaudhary
Dr. Abhijeet Botre [MBBS, DCH, DNB (Paediatric Neurology & Epilepsy)]
Dr. Abhijeet Kumar
Dr. Abhijeet Palshikar [MBBS, MD - General Medicine, DNB - Cardiology]
Dr. Abhijeet Palshikar [MBBS, MD - Medicine, DNB - Cardiology, FESC, FACC, FSCI]
Dr. Abhijeet Wahegaonkar [MBBS, D(Ortho), DNB (Ortho), M.Ch (Ortho), MS-CTSA (Mayo Clinic), FACS, Diplomate in Hand Surgery]
Dr. Abhijit A Bagde [MBBS, DNB - Pediatrics, Fellowship - Pediatric Intensive Care]
Dr. Abhijit Agashe [MBBS, MS, DNB, FASIF, Fellow Joint Replacement Surgery]
Dr. Abhijit Bagade [MBBS, MD - General Medicine]
Dr. Abhijit Taraphder
Dr. Abhijit Vaidya
Dr. Abhimanyu
Dr. Abhinandan Mishra
Dr. Abhinandan Mukhopadhyay [MBBS, MS - General Surgery, MCh - Urology/Genito-Urinary Surgery]
Dr. Abhinav Aggarwal
Dr. Abhinav Agrahari
Dr. Abhinav Gupta [MBBS, MD, DM - Neurology]
Dr. Abhinav Jain
Dr. Abhinav Nair [MBBS, MD (Internal Medicine)]
Dr. Abhinav Narwariya [MBBS, MD - Radiotherapy, DNB - Medical Oncology]
Dr. Abhinav Sharma
Dr. Abhinit Kumar
Dr. Abhinit kumar
Dr. Abhishek Agrawal
Dr. Abhishek Deepak [MBBS, MD - General Medicine, DM - Gastroenterology]
Dr. Abhishek Deo
Dr. Abhishek Goel
Dr. Abhishek Gupta
Dr. Abhishek Kathuria [DM-Gastroenterology, MD -Medicine, MBBS]
Dr. Abhishek Kumar Gupta [BPT - Jyoti College of Physiotherapy Bareilly, MPT]
Dr. Abhishek Malviya [MBBS, Diploma - Otorhinolaryngology, DNB]
Dr. Abhishek Nandi
Dr. Abhishek Patil [MBBS, MD - Medicine, DNB - Rheumatology]
Dr. Abhishek Raj
Dr. Abhishek Rathore
Dr. Abhishek Raval [MBBS, MD General Medicine, DM Cardiology]
Dr. Abhishek Singh [MBBS, DLO, DNB - ENT]
Dr. Abhishek Srivastava [MBBS, MS - Orthopaedics, Clinical Fellow - Spine Surgery]
Dr. Abhishek Vaish [MBBS, MS (Ortho), MCh (Ortho), DNB (Ortho), Dip. SICOT (Rome), MNAMS]
Dr. Abraham Kurien
Dr. Abrar Ahmed
Dr. Achal Bhagat [MBBS,MD, MRCP]
Dr. Achint Krishna
Dr. Achintya Sharma [MBBS, MS in General Surgery, MCh in Vascular Surgery]
Dr. Adarsh Chaudhary
Dr. Adhish Basu
Dr. Adhishwar Sharma
Dr. Adhishwar Sharma
Dr. Aditi Aggarwal [MBBS, PDCR, MD - Radiation Oncology]
Dr. Aditi Chaturvedi
Dr. Aditi Chopra [MBBS, MD - Medicine, DM - Endocrinology]
Dr. Aditi Kaskar
Dr. Aditi Krishna
Dr. Aditya Aggarwal [MBBS, MS - General Surgery, MCh - Plastic Surgery, DNB - Plastic Surgery]
Dr. Aditya Dixit [MBBS, MD - Pediatrics]
Dr. Aditya Gupta [MBBS, MS, FMAS - General Surgery]
Dr. Aditya Gupta [Specialist Cyberknife RadioSurgery ]
Dr. Aditya Jain
Dr. Aditya Kulkarni [MS, DNB, FRCS General Surgery, MCh Surgical Gastroenterology]
Dr. Aditya Mantry
Dr. Aditya Nanavati
Dr. Aditya Singh Bhati
Dr. Adosh Lall
Dr. Adulkar Deepanjali G
Dr. Advait Kothurkar [MS, DNB (General Surgery), MRCS]
Dr. Adwaita Gore
Dr. Aftab Khan
Dr. Agneesh Patial
Dr. Agni Sekhar Saha
Dr. Agnivesh Tikoo [MBBS, MS - Orthopedics, AO Spine Fellowship]
Dr. Agya Singh Kochar
Dr. Ahmar Nauman Tarique
Dr. Ahmed Kamaal
Dr. Ahmed Shatila [Neurologist, Multiple Sclerosis]
Dr. Aishwarya Shukla
Dr. Ajay Doiphode
Dr. Ajay Gupta
Dr. Ajay Kaul [MBBS, MS - General Surgery, MCh - Cardiothoracic Surgery]
Dr. Ajay Khanna
Dr. Ajay Kr Arya
Dr. Ajay Kumar
Dr. Ajay Kumar Ajmani
Dr. Ajay kumar Chauhan [MBBS, DNB - General Surgery, FICS]
Dr. Ajay Kumar Gupta
Dr. Ajay Kumar Kriplani
Dr. Ajay Kumar Sharma
Dr. Ajay Lall
Dr. Ajay Naik [MS, MCh]
Dr. Ajay Sharma [MBBS, MD - Medicine, DNB - Medical Oncology]
Dr. Ajaya Kashyap
Dr. Ajit Dandekar [MBBS, MD, Diploma - Psychiatric Medicine]
Dr. Ajit Kumar Roy [MBBS, MD - Medicine, DM - Neurology]
Dr. Ajit Saxena [MBBS, MS, DU]
Dr. Ajit Singh Baghela [ DNB - Paediatric Neurologist ]
Dr. Ajit Singh Narula
Dr. Ajit Singh Narula [MBBS, MD - General Medicine, DM - Nephrology]
Dr. Ajitabh Srivastava [MBBS, MS - General Surgery, DNB - General Surgery]
Dr. Ajitha Kumari K [Pediatric Cardiology]
Dr. Aju Jacob [MBBS, MS - General Surgery, MCh - Surgical Gastroenterology]
Dr. Aju Jacob [MBBS, MS - General Surgery, MCh - Surgical Gastroenterology]
Dr. Akanksha Chichra [MBBS, DNB - Paediatrics, FNB - Pediatric Hematology Oncology]
Dr. Akash Bhayana [MBBS, MD - Skin & VD]
Dr. Akash Gupta
Dr. Akash Khandelwal [MBBS, MD - General Medicine, DM - Hematologist]
Dr. Akhil Govil
Dr. Akhilesh Rathi
Dr. Akhter Jawade
Dr. Aklesh Tandekar [MBBS, MD, Fellowship - Critical Care]
Dr. Akshat Malik
Dr. Akshatha Sharma [MS(ObGyn),MRCOG,Fellowship in Fetal Medicine]
Dr. Akshay Budhraja
Dr. Akshay Chhallani [MBBS, Fellowship - Internal Medicine, DNB - Internal Medicine]
Dr. Akshay K Mehta
Dr. Akshay Tiwari
Dr. Akshit Gupta
Dr. Alagu Balaji P
Dr. Ali Irani
Dr. Alisha Chaubal
Dr. Alka Bhasin
Dr. Alka Dahiya [MBBS, MD - Obstetrics & Gynaecology, MCh - Gynaecological Oncology]
Dr. Alka Gupta Upchaar
Dr. Alka Kumar
Dr. Alka Pandey [MBBS and an MS in Ophthalmology]
Dr. Alka Sinha
Dr. Alkesh Jain
Dr. Alok Gupta [DM - Oncology, MD - General Medicine, MBBS]
Dr. Alok Kumar Agarwal [MBBS, MD, MRCP(UK)]
Dr. Alok Kumar Pandey [MBBS , MS General Surgery, DRNB Gastrointestinal Surgery, Fellowship in Liver Transplant and Hepatobiliary Surgery]
Dr. Alok Narang [MBBS, MS (Gen Surg)]
Dr. Alok Tiwari [DNB - Surgical Oncology, MS - General Surgery, MNAMS-General Surgery]
Dr. Alokananda Das
Dr. Aloy J Mukherjee
Dr. Amal Roy Chaudhoory
Dr. Aman Dhillon
Dr. Aman Dua
Dr. Aman Gupta [MBBS, MS - General Surgery, MCh - Urology]
Dr. Amandeep Gujral
Dr. Amanjeet Singh [DNB - Surgical Gastroenterology, MS-General Surgery, MBBS]
Dr. Amar Bist
Dr. Amar Nath Ghosh
Dr. Amar Pal Singh Suri
Dr. Amar Shinde
Dr. Amardeep Yadav
Dr. Amarender Singh Puri
Dr. Ambalika Veer [MD Nuclear Medicine]
Dr. Ambar Khaira
Dr. Ambrish Mithal
Dr. Ameet Kishore
Dr. Ameet Kumar Mandot [MBBS, MD - Internal Medicine, DNB - Gastroenterology]
Dr. Amey Sonavane [MBBS, DNB- General Medicine, DNB - Gastroenterology]
Dr. Amieleena Chhabra
Dr. Amish Chaudhary
Dr. Amish Mhatre
Dr. Amit Agarwal
Dr. Amit Ashok Bhatti
Dr. Amit Bansal [MBBS, MS - General Surgery, MCh - Urology/Genito-Urinary Surgery]
Dr. Amit Bhargava [MBBS, Diploma - Radiology Therapy, DNB - Oncology]
Dr. Amit Bhatt [European Certification in Medical Oncology (ESMO), Masters in Molecular Oncology (CNIO, Spain)]
Dr. Amit Chandra
Dr. Amit Chaudhry
Dr. Amit Dubey [MBBS, Diploma - Anaesthesia]
Dr. Amit Dutt Dwary
Dr. Amit Ghose
Dr. Amit Goel [MBBS, DNB - Critical Care]
Dr. Amit Gupta [MBBS, MD - Medicine, DNB - Nephrology, FRCP-London]
Dr. Amit Jain
Dr. Amit Kapoor [MBBS, DNB - Neurosurgery, Diploma - Accident & Emergency Medicine]
Dr. Amit Kumar
Dr. Amit kumar Agarwal
Dr. Amit Kumar Chaurasia [MBBS, MD - Medicine, DM - Cardiologist]
Dr. Amit Kumar Mahapatray
Dr. Amit Kumar Ray
Dr. Amit Kumar Singhal
Dr. Amit Kumar [MBBS, MD - Medicine, DNB - Cardiology]
Dr. Amit Langote [MBBS, MD - Internal Medicine, DNB - Nephrology]
Dr. Amit Miglani [MBBS, MD - General Medicine, DM - Gastroenterology]
Dr. Amit Misri
Dr. Amit Mittal [MBBS, MD - General Medicine, DM - Gastroenterology]
Dr. Amit Mittal [MBBS, MD - Internal Medicine, DM - Gastroenterology]
Dr. Amit Nath Rastogi
Dr. Amit Pandita [MBBS, MD (General Medicine), DNB (Medical Gastroenterology)]
Dr. Amit Pendharkar [MBBS, MD - Medicine, DM - Cardiology]
Dr. Amit Rane [MBBS, DNB - General Surgery, DrNB Vascular Surgery]
Dr. Amit Rauthan [ [MBBS, MD - Internal Medicine, DM - Medical Oncology]]
Dr. Amit Srivastava
Dr. Amit Valbhani [MBBS, MD - Paediatrics]
Dr. Amit Vatkar
Dr. Amit Vij
Dr. Amita Jain
Dr. Amita Mahajan [MBBS, MD, MRCP]
Dr. Amita Naithani [MBBS, Diploma - Gynaecology and Obstetrics, DNB - Obstetrics And Gynecology]
Dr. Amita Wadhwa [MBBS, MD - Obstetrics & Gynaecology, DNB - Obstetrics & Gynecology]
Dr. Amitabh Singh
Dr. Amitabh Singh [MBBS, MS - General Surgery, MCh - Plastic Surgery, International Fellowship]
Dr. Amitabha Dutta [MBBS, FRCP]
Dr. Amitabha Ghosh
Dr. Amitava Pahari
Dr. Amitesh Chakravarty
Dr. Amith K Pakkala
Dr. Amod Patankar [MDS]
Dr. Amogh R Vaishampayan
Dr. Amol Balvant Akhade
Dr. Amol Bansode [FCPS, DNB (Medicine)]
Dr. Amol Dumbre [DNB General Surgery]
Dr. Amol Joshi
Dr. Amol kumar Patil [MBBS, MS - General Surgery, DNB - Urology]
Dr. Amol Patil
Dr. Amol Talulikar [MBBS, MS (General Surgery), FRCS (Urology)]
Dr. Amol Vadgaonkar
Dr. Amrish Kumar
Dr. Amrita Kapoor Chaturvedi
Dr. Amrita Rao [MBBS, DNB - Obstetrics & Gynaecology]
Dr. Amrita Roy
Dr. Amrita Srivastava
Dr. Amritendu Mukherjee
Dr. Amudhan A
Dr. Amvrin Chatterjee
Dr. Anadi Pachaury [MBBS, MS - General Surgery, MCh - Surgical Oncology]
Dr. Anagha Behere [DNB (Ophthalmology), DOMS]
Dr. Anaita Udwadia Hegde [MBBS, Diploma - Child Health, MD - Pediatrics ,Fellowship - Pediatric Neurology]
Dr. Anand Alurkar [MD, DM (Neurology)]
Dr. Anand Dotihal [MD - Medicine, DM - Gastroenterology]
Dr. Anand Gupta [MBBS, MD - PGI Chandigarh]
Dr. Anand Jaiswal
Dr. Anand Katkar [MBBS, DNB, MS]
Dr. Anand Kumar
Dr. Anand Kumar Saxena
Dr. Anand Narayanan [MBBS, MD - Paediatrics]
Dr. Anand P [MBBS, MD - Paediatrics, DNB, EULAR Fellow]
Dr. Anand Patil [MBBS, MS, DrNB - Urology]
Dr. Anand Prakash
Dr. Anand Praveen Kumar A
Dr. Anand R Shenoy
Dr. Anand Sinha
Dr. Anand Subramanyam
Dr. Anandh Balasubramaniam
Dr. Anant Kumar
Dr. Anantheswar Y N [MBBS, MS - General Surgery, MCh - Plastic Surgery]
Dr. Ananya Das [MBBS, MD - Internal Medicine, PGDGM - Geriatric Medicine]
Dr. Anchit Uppal [MBBS, DNB, MCh - Orthopaedics]
Dr. Aneesh Karwa [MBBS, MD, DrNB - Nephrology]
Dr. Aneesh Sabnis
Dr. Angshuman Das
Dr. Aniel Malhotra [MBBS, MS, DOMS]
Dr. Aniket Deshmukh
Dr. Aniket Joshi [MBBS, FCPS (Medicine)]
Dr. Anil Bhan
Dr. Anil D Cruz
Dr. Anil Dcruz [MBBS, MS, DNB - Surgical Oncology]
Dr. Anil Dhall [MBBS, MD - Medicine, DM - Cardiology, FACC, FESC, FSCAI, FCSI]
Dr. Anil Godbole
Dr. Anil Handoo
Dr. Anil Karadkar
Dr. Anil Karkhanis
Dr. Anil Krishna Gundala
Dr. Anil Kumar Anand
Dr. Anil Kumar B T [MBBS, DNB - Nephrology]
Dr. Anil Kumar Behl
Dr. Anil Kumar Gulia [MBBS, MS - General Surgery, MCh - Urology]
Dr. Anil Kumar Gupta [MBBS, MD - Internal Medicine]
Dr. Anil Kumar Jain
Dr. Anil Kumar Kansal [MBBS, MS - General Surgery, MCh - Neurosurgery]
Dr. Anil Kumar Murarka
Dr. Anil Mandhani
Dr. Anil Potdar
Dr. Anil Saxena [MBBS, MD - Medicine, DNB - Cardiology]
Dr. Anil Sharma [MBBS, Mch - Urology, Fellowship - Renal transplant]
Dr. Anil Thakwani [MBBS, MD - Radiotherapy]
Dr. Anil Venkitachalam
Dr. Anil Venkitachalam
Dr. Animesh Saha
Dr. Anirban Deep Banerjee
Dr. Anirban Dutt
Dr. Aniruddh Trivedi [MBBS, MS - General Surgery, MCh - Thoracic Surgery]
Dr. Aniruddha Bhosale [MBBS, DNB (General Surgery), FMAS, MRCS (UK), FACS (USA), Fellowship in Liver Transplant & HPB Surgery]
Dr. Aniruddha Deshpande [MS Orthopaedics]
Dr. Aniruddha Vidyadhar Kulkarni
Dr. Anish Garg [MBBS, MD, DM - Nephrology]
Dr. Anish Garg [MBBS, MD, DM - Nephrology]
Dr. Anish Gupta [MBBS, MS (ENT and Head & Neck Surgery]
Dr. Anish Nandkumar Tawde
Dr. Anita Bakshi [MBBS, MD]
Dr. Anita Jatana [PG (Dietetics)]
Dr. Anita Kaul [MBBS, MS, MRCOG FRCOG ,FICOG ,Dip in Fetal Medicine UK,Dip in Advanced Obstetric Scanning UK]
Dr. Anita Malik
Dr. Anita Maria Dias [MBBS, MD - General Medicine]
Dr. Anita Mathew
Dr. Anita Pramod [MBBS, DNB - Anaesthesiology]
Dr. Anita Saxena
Dr. Anita Sethi [MD - Ophthalmology, MBBS, DNB - Ophthalmology]
Dr. Anitha Kumari A M [MBBS, DLO, DNB - ENT]
Dr. Anjali Bhosle
Dr. Anjali Gupta
Dr. Anjali Kelkar [DCP, DNB Pathology]
Dr. Anjali Patki
Dr. Anjan Bhattacharya
Dr. Anjana B Choudhury [MBBS, MD - Paediatrics]
Dr. Anjana Satyajit [Head - Dentistry | Bachelor of Dental Surgery]
Dr. Anjani Kumar Agrawal
Dr. Anjila Aneja
Dr. Anju Mangla [MBBS, MD]
Dr. Anju Singh [MBBS, Diploma in Child Health (DCH), MD - Pediatrics | Consultant Pediatric Rheumatologist]
Dr. Ankit Dalal
Dr. Ankit Garg
Dr. Ankit Jain [MBBS, MS, MCh, FINS, FINR]
Dr. Ankit Kedia
Dr. Ankit Mahuvakar
Dr. Ankit Parakh
Dr. Ankit Shah [BDS, MDS - Oral & Maxillofacial Surgery]
Dr. Ankita Baidya
Dr. Ankur Arya [MBBS, MS - General Surgery, MCh - Urology/Genito-Urinary Surgery]
Dr. Ankur Atal Gupta
Dr. Ankur Bahl
Dr. Ankur Bhatnagar [MBBS, MS, MCh - Urology]
Dr. Ankur Garg [MBBS, MS - General Surgery, MCh - Hepatobillary Surgery and Liver Transplant]
Dr. Ankur Jain
Dr. Ankur Jain
Dr. Ankur Kumar Jindal [MBBS, MD - Pediatrics, DM - Pediatric Clinical Immunology and Rheumatology]
Dr. Ankur N Gupta
Dr. Ankur Pruthi
Dr. Anmol Maria
Dr. Anoop Amarnath
Dr. Anoop Bandil [MBBS, DNB]
Dr. Anoop Kohli
Dr. Anoop Purkayastha
Dr. Anshu Mahajan
Dr. Anshul Goel
Dr. Anshul Gupta [MBBS, MD - General Medicine, DM - Oncology]
Dr. Anshul Jain
Dr. Anshumali Mishra
Dr. Anshuman Agarwal
Dr. Anshuman Kaushal [MS - General Surgery, MBBS]
Dr. Anshuman Kumar [MBBS, MS - General Surgery, MCh - Onco Surgery, Robotic Oncosurgeon ]
Dr. Anshuman Manaswi
Dr. Anshuman Singh [MBBS, MD - Internal Medicine, DM - Gastroenterology]
Dr. Anu Gupta [MBBS, MS - Obstetrics & Gynaecology]
Dr. Anu Prasad G
Dr. Anubha Mittal
Dr. Anubhav [MBBS, MS - General Surgery, MCh - HPB Surgery, Liver Transplant Fellowship]
Dr. Anuj Chawla
Dr. Anuj Pahuja
Dr. Anuj Sathe [MBBS, MD - Internal Medicine, DM - Cardiology]
Dr. Anuja Athalye [DNB General Surgery]
Dr. Anuja Kulkarni [DNB (General Medicine)]
Dr. Anuja Lakra [MBBS, MD - Internal Medicine]
Dr. Anuja Pethe
Dr. Anuja Porwal [MBBS, DNB - Internal Medicine, DNB - Nephrology]
Dr. Anuja Thomas [MBBS, DGO, DNB - Obstetrics & Gynaecology]
Dr. Anunaya Katiyar [MBBS,MD PEDIATRICS, FELLOW IN PEDIATRICS NEPHROLOGY]
Dr. Anup Bansode [MBBS, MS (Orthopaedics)]
Dr. Anup Chaudhari
Dr. Anup Dhir
Dr. Anup Gulati [MBBS, MS - General Surgery, DNB - Urology]
Dr. Anup Kumar Thacker
Dr. Anup P Nair [MBBS, MS - General Surgery, MCh - Neurosurgery]
Dr. Anup Prakash Rawool [DM-Medical Genetics, MD-Internal Medicine, MBBS]
Dr. Anup Prakash Rawool [MBBS, MD (Internal Medicine), DM (Medical Genetics), MBA (Hospital Administration), PGDGM, PGDHHM, CCMG]
Dr. Anupam Bahl [MBBS DNB(General Medicine) DNB(Nephrology)]
Dr. Anupam Bhargava [MBBS, MS, Diploma - Urology]
Dr. Anupam Chakrapani
Dr. Anupam Das [MBBS, MS, MCh - CTVS, FRCS, FACS]
Dr. Anupam Goel
Dr. Anupam Kumar Sharma [MBBS, MS(General Surgery), DNB(Urology)]
Dr. Anupam Roy
Dr. Anupam Saha
Dr. Anupam Sibal [MD, FIAP, FIMSA, FRCP(Lon), FRCP(Glas), FRCPCH, FAAP]
Dr. Anupama Mane [MBBS, MS - General Surgery, Fellowship - Breast Oncosurgery]
Dr. Anuradha Ghorpade
Dr. Anuradha Vinod [MBBS, DCH, DNB - Paediatrics]
Dr. Anurag Krishna
Dr. Anurag Mahale
Dr. Anurag Passi
Dr. Anurag Saxena
Dr. Anurag Sharma
Dr. Anurag Sharma [MBBS, MS - General Surgery, DNB - Neurosurgery]
Dr. Anurag Singh [BDS, MDS - Oral & Maxillofacial Surgery and Oral Implantology]
Dr. Anusha Katare [MBBS, MD - Dermatology]
Dr. Anusha Reddy Badvelli
Dr. Anusheel Munshi [MBBS, MD - Radiotherapy]
Dr. Anushtup De
Dr. Anutrisha Bhaumik
Dr. Anvay Mulay [MBBS, MS - General Surgery, MCh - Thoracic Surgery]
Dr. Anwin J
Dr. Aparna Chakravarty
Dr. Aparna Govil Bhasker [MBBS, MS - General Surgery, Fellowship - Advanced Laparoscopic & Bariatric Surgery]
Dr. Aparna H Mahajan [MBBS, MS-ENT, MRCS- ENT]
Dr. Aparna Jaswal
Dr. Aparna Katyal [MBBS, MD - Radiology]
Dr. Apurva Arora
Dr. Aradhana Chauhan [MBBS, DNB (General Medicine), DNB (Neurology)]
Dr. Arati Shahade [MD (General Medicine), PG Diploma (Diabetes)]
Dr. Archana
Dr. Archana Elangovan
Dr. Archana G. Mahajan
Dr. Archana K. Raichurkar [MBBS, MD - Anaesthesiology, DM - Neuroanaesthesia]
Dr. Archana M [MBBS, DNB - Paediatrics, Fellowship in Paediatric Infectious Diseases]
Dr. Archana Ranade
Dr. Archana S [BDS, MDS, Cert. in Implantology]
Dr. Archana Yadav [MBBS, MD - Paediatrics]
Dr. Archit Pandit [MBBS, Gold Medalist MS - General Surgery, Fellowship - Thoracoscopic Lung Cancer Surgery]
Dr. Arif Wahab [MD (Medicine), DM (Cardiology)]
Dr. Arijit Chattopadhyay
Dr. Arindam Mondal
Dr. Arindam Mukherjee
Dr. Aritra Konar
Dr. Arjun Goel [MBBS, MS, FNB - Minimal Access Surgery]
Dr. Arjun Khanna
Dr. Arjun Verma [MBBS, MD - Anaesthesiology, IDCCM, IFCCM]
Dr. Arnab Basak
Dr. Arnab Dasgupta [MBBS, MD - Anaesthesiology]
Dr. Arpan Chakraborty
Dr. Arpan Dev Bhattacharya [MBBS, MD, DM, MRCP, FRCP (Edinburgh)]
Dr. Arpit Jain [MBBS, MD - Internal Medicine]
Dr. Arpit Verma [MBBS, MS General Surgery, MCh. Plastic Surgery]
Dr. Arpita Roy Choudhury
Dr. Arti Pawaria
Dr. Aru Chhabra Handa
Dr. Arun Behl
Dr. Arun Bhardwaj
Dr. Arun Chowdary Kotaru [MD - Pulmonary Medicine, DNB - Respiratory Medicine, MBBS]
Dr. Arun Dewan
Dr. Arun Fernandes [MS (General Surgery)]
Dr. Arun Garg
Dr. Arun Garg
Dr. Arun Kumar Giri
Dr. Arun Kumar Sundaram
Dr. Arun Saroha [MCh - Neuro Surgery, MBBS]
Dr. Arun Sharma [BDS, MDS-Oral and Maxillofacial Surgery]
Dr. Aruna Bhave
Dr. Aruna Kumar [MBBS, DVD, DNB - Dermatology]
Dr. Aruna Rajendran [MBBS, MD - Obstetrics & Gynaecology]
Dr. Aruna Rajendran [MBBS, MD - Obstetrics & Gynaecology]
Dr. Arunabha Sengupta
Dr. Arundhati De
Dr. Arundhati Kale Sidhaye [MBBS, DNB (Ophthalmology), DOMS]
Dr. Arup Sahu
Dr. Arvind Bagga [MD, FAMS, FIAP, FISN]
Dr. Arvind Dambalkar [MBBS, MD, DM - Cardiology]
Dr. Arvind Sethi
Dr. Arvinder Singh Soin
Dr. Arwa Mohsin E [MBBS, MD - Obs & Gyn]
Dr. Asad Riyaz
Dr. Asawari Kesari Kapoor [M.B.B.S, D.G.O(Mumbai) ,D.G.O (C.P.S), D.N.B (OBGY)]
Dr. Aseem R. Srivastava[MBBS, MS - General Surgery, MCh - Cardio Thoracic Surgery | Paediatric Cardiac Surgery & Paediatric Cardiology]
Dr. Asfar Ali
Dr. Asgar Ali [MBBS, MS - Orthopaedics, Fellowship in Spine Surgery]
Dr. Asha Sharma [Gynecologist,Obstetrician and Laparoscopic Surgeon ]
Dr. Ashfaque Ahmed
Dr. Ashiq Ali Raval
Dr. Ashish Agarwal
Dr. Ashish Chakravarty
Dr. Ashish Chauhan
Dr. Ashish Dixit [MBBS, MD, DNB, DM - Clinical Haematology]
Dr. Ashish George
Dr. Ashish Gupta
Dr. Ashish Gupta
Dr. Ashish Gupta [MBBS, Medical Oncology )
Dr. Ashish Jai Kishan
Dr. Ashish Kakar [BDS,MDS]
Dr. Ashish Katewa
Dr. Ashish Khanijo [MBBS, MS General Surgery, DNB Cardiothoracic Surgery]
Dr. Ashish Khattar
Dr. Ashish Nandwani
Dr. Ashish Philip Zachariah
Dr. Ashish Sabharwal
Dr. Ashish Sharma
Dr. Ashish Singhal
Dr. Ashish Tomar
Dr. Ashish Vashishth [MBBS, MS - ENT, DNB]
Dr. Ashok Bhanage [MBBS, MS General Surgery, MCh Neuro Surgery]
Dr. Ashok Damir
Dr. Ashok Dutta [MD]
Dr. Ashok Gawdi [MBBS, MD - Pediatrics]
Dr. Ashok K Khera
Dr. Ashok Kumar Agarwal [MD - General Medicine, MBBS]
Dr. Ashok Kumar Vaid
Dr. Ashok Omar
Dr. Ashok Rajagopal
Dr. Ashok Rajput [MBBS, Diploma - TB and Chest Disease, MD - Chest Medicine]
Dr. Ashok Rijhwani
Dr. Ashok Sarin [MBBS, MD, FRCP(Edin)]
Dr. Ashok Sengupta
Dr. Ashok Sharma
Dr. Ashok Singhvi
Dr. Ashu Abhishek
Dr. Ashu Agarwal [MBBS, MS]
Dr. Ashu Consul
Dr. Ashutosh Chavan
Dr. Ashutosh Dudhatra
Dr. Ashutosh Gupta
Dr. Ashutosh Marwah [MBBS, MD Pediatrics, Fellowship in Pediatric Cardiology]
Dr. Ashutosh N. Shetty
Dr. Ashutosh Shrivastava [MBBS, MS - Orthopaedics, Fellowship in Arthroscopy – Singapore]
Dr. Ashutosh Shukla
Dr. Ashutosh Yadav [MBBS, MD (Medicine), DM-Cardiology , Consultant - Interventional Cardiology]
Dr. Ashwani Kumar Sharma [MBBS, MS - General Surgery, DNB - Surgical Oncology, Robotic Surgeon]
Dr. Ashwani Kumar Singh
Dr. Ashwathy Haridas [MBBS, MD - General Medicine, DM - Nephrology]
Dr. Ashwin Sunil Tamhankar [MBBS, MS - General Surgery, Mch - Urology]
Dr. Ashwini Annam
Dr. Ashwini Goel
Dr. Ashwini Nagarkar [MBBS, DNB ENT]
Dr. Asif Iqbal
Dr. Asim Kumar Kandar
Dr. Asit Arora
Dr. Asmit Vaidya [MBBS, MD (Internal Medicine)]
Dr. Asmita Potdar [MBBS, DNB - Obstetrics & Gynaecology]
Dr. Astha Gupta [MBBS, MD - Dermatology, V.D.]
Dr. Astha Sharma [MBBS, MD, Fellowship - Aesthetics and Lasers]
Dr. Atanu Jana
Dr. Ateksha Bhardwaj Khanna
Dr. Athar Parvez Ansari [MBBS, MD - Internal Medicine]
Dr. Atma Ram Bansal
Dr. Attawar Sandeep
Dr. Attique Vasdev
Dr. Atul Ahuja [MBBS ,MS(ENT)]
Dr. Atul D. Sajgure [MBBS, MD (Medicine), DNB (Nephrology), DM (Nephrology)]
Dr. Atul Joshi [MD (Physician, USSR)]
Dr. Atul Kumar Mittal [MBBS, MS - ENT and Otorhinolaryngology]
Dr. Atul Limaye
Dr. Atul Luthra
Dr. Atul Mathur
Dr. Atul Mishra [MBBS, MS - Orthopedics, Fellowship - Knee Orthopedics & Sports Injury]
Dr. Atul Morarji Ganatra
Dr. Atul Prasad
Dr. Atul Seth [MBBS, DO, DNB - Ophthalmology]
Dr. Atul Sharma
Dr. Atul Sharma [MBBS, MS, DNB, MCh - Plastic Surgery]
Dr. Atul Taneja
Dr. Atul Verma
Dr. Atul Vinayak Ingle
Dr. Atul Vinayak Ingle
Dr. Atul Wankhede
Dr. Avanish Arora
Dr. Avanish Saklani
Dr. Avdhesh Bansal [MBBS,MD, MRCP]
Dr. Avi Kumar
Dr. Avinash Agarwal [MBBS, DNB, MCh, Cosmetic & Plastic Surgery]
Dr. Avinash Kumar Singh
Dr. Avinash Mani
Dr. Avinash Verma
Dr. Avinash Walawalkar
Dr. Avishek Poddar
Dr. Avnindra Gupta
Dr. Avnish Kumar Seth [MBBS, MD - General Medicine, DM - Gastroenterology]
Dr. Avtar Singh Bath [MBBS, MS - General Surgery, MCh - Plastic Surgery]
Dr. Ayaz Ahmed [MBBS, MD - Internal Medicine, DNB - Cardiology]
Dr. Ayush Chawla [MBBS, DNB, DOHNS - ENT]
Dr. Ayush Gupta
Dr. Azeez Pasha [MBBS, MS, FVES, FACS, FRCS (Glasgow)]
Dr. Azhar Perwez [MBBS, MS - General Surgery, DNB - Surgical Gastroenterology]
Dr. Azmat Karim
Dr. B Anand Shankar [MBBS, MD - Anaesthesiology]
Dr. B Anand Shankar [MBBS, MD - Anaesthesiology]
Dr. B D Pathak [MBBS, MS - General Surgery]
Dr. B K Singhania
Dr. B M L Kapoor [MBBS, MS, FRCS, FAMS]
Dr. B Niranjan Naik
Dr. B Radhika
Dr. B Y T Arya [MD, DRM]
Dr. B. A. Chandramouli
Dr. B. Kiran Kumar [MBBS, MD - Paediatrics, Fellowship - Paediatric Intensive Care]
Dr. B. S. Murthy [MBBS, MS - Orthopaedics, Fellowship - Join Replacement]
Dr. B. S. Ramakrishna
Dr. Balachandra B V [MBBS, MD, FPCC, DAA]
Dr. Balaji Dhandapani
Dr. Balaji Dhandapani [MBBS, MS, M.Ch (Plastic Surgery)]
Dr. Balasubramaniam Govini [MS - General Surgery, MBBS, MCh - Cardio Thoracic Surgery]
Dr. Balasubramanyam Shankar [MD, FRCR, EDiR]
Dr. Balbir Singh [MBBS, MD - Internal Medicine, DM - Cardiology]
Dr. Baliwanth A [MBBS, DCH, DNB, MRCPCH (UK)]
Dr. Balkishan Gupta [MBBS, MS - General Surgery]
Dr. Balvinder Rana
Dr. Bapujee N Sawant
Dr. Basade Siraj [MS, MCh Neurosurgery]
Dr. Bashi V. Velayudhan
Dr. BELA KEDIA
Dr. Bela Makhija
Dr. Belal Bin Asaf
Dr. Bhaba Nanda Das [MBBS, MS, MCh]
Dr. Bhabatosh Biswas
Dr. Bhagwantsingh Zorawer Singh Ratta [MBBS, MS, and Dip.Paediatric Surgery ]
Dr. BHAGWAT CHOUDHARY
Dr. Bhagwat Narayan Rajput [MBBS, DNB - Psychiatry, MD - Psychiatry]
Dr. Bhanu Madan [BDS, MDS - Maxillofacial Prosthodontics]
Dr. Bhanu Prasad [MBBS (UK), MRCEM (UK)]
Dr. Bharat Agarwal [MBBS, DNB - General Medicine]
Dr. Bharat Bahre
Dr. Bharat Bhushan Chauhan [MBBS, MS - Orthopaedics, Fellowship]
Dr. Bharat Patel
Dr. Bharat Rajivkumar Saxena
Dr. Bharat Rawat
Dr. Bharat Shah [MBBS, MD - Psychiatry]
Dr. Bharath Kadadi [MBBS, MS - Orthopaedics]
Dr. Bharati Rajguru [MA Psychology, PhD Psychology]
Dr. Bhaskar BV [MBBS, MS, MCh - CTVS]
Dr. Bhaskar Nandi
Dr. Bhaskar Pal
Dr. Bhaskar Viswanathan
Dr. Bhavi Barchha
Dr. Bhavisha Ghugare
Dr. Bhavna Anand [MBBS, MS, Fellowship]
Dr. Bhavna Banga
Dr. Bhawna Attri [MBBS, MD, DM - Endocrinology]
Dr. Bhawna Saddy Awasthy
Dr. Bheem Raj Gupta [MBBS, MD - Medicine, DNB - Nephrology]
Dr. Bhooshan Zade [MD Radiotherapy]
Dr. Bhumi Dave [MBBS, MD Medicine, IDC]
Dr. Bhumika Madan
Dr. Bhupendra Chaudhary [MBBS, MD - Counselling and Psychotherapy]
Dr. Bhupesh Singh
Dr. Bhushan Chavan [MBBS, DNB, Fellowship - Pediatric Cardiology]
Dr. Bhushan Khedkar [MBBS, DNB Orthopaedics]
Dr. Bhushan Nariani [MBBS, DePuy Fellowship - Arthroplasty, MS - Orthopedics, Robotic knee replacement surgeon ]
Dr. Bhuvan Chanana
Dr. Bhuvan Chugh
Dr. Bhuvnesh Kumar Aggarwal
Dr. Bidisha Banerjee [MBBS, MD, DM - Paediatric Neurology]
Dr. Bijaylaxmi Behera [MBBS, MD - Pediatrics, DM - Neonatology]
Dr. Bijendra Kumar Sinha [MBBS, MS - General Surgery]
Dr. Bijoy Kumar Nayak [MD - Obstetrics & Gynaecology, MBBS]
Dr. Bikas Bhattacharya
Dr. Bikash Majumder
Dr. Bikram k Mohanty
Dr. Bikram Kumar Deka
Dr. Bilal Ahmad Baba [MBBS, MD - General Medicine, DM - Cardiology]
Dr. Bimal Kumar Sahu [MBBS, MD - General Medicine, DM - Gastroenterology]
Dr. Bindu K S [MBBS, MD, DGO]
Dr. Binit Kedia [MBBS, MS, MCh - Neurosurgery]
Dr. Bipin Kumar Dubey [MBBS, MD - Medicine, DM - Cardiology]
Dr. Bipin Walia
Dr. Biplab Das
Dr. Bipul Kumar Sinha
Dr. Bir Singh Sehrawat
Dr. Biswajit Sengupta
Dr. Biswajyoti Hazarika [MBBS, MS - ENT, Fellow Surgical Oncology, Robotic Head & Neck Surgery
Dr. Biswanath Mukopadhyay
Dr. Biswanath Roy
Dr. Biswaroop Mukherjee
Dr. Bivas Biswas
Dr. Blossom Dsouza
Dr. BML Kapoor
Dr. Boman Nariman Dhabhar
Dr. Boopathy John
Dr. Bopanna K M
Dr. Brajesh Kumar Koushle
Dr. Brig Parthasarathi Moulick
Dr. Buchun Mishra
Dr. Buddhadeb Chatterjee
Dr. C Vijay Bose
Dr. C. Nivedita
Dr. C.M Malhotra
Dr. C.S. Agarwal
Dr. Carreen Pakrasi
Dr. Chaitali Trivedi Mahajan
Dr. Chakor Vora [MD General Medicine, DM Medical Oncology]
Dr. Chakrapani B S
Dr. Chanchal Pal [MS ( ENT and Head & Neck Surgery ) MBBS]
Dr. Chanda Chowdhury
Dr. Chandan Arora [MBBS, MS - Orthopaedics]
Dr. Chandar Mohan Batra [MBBS, DCH, MD (Internal Medicine), DNB (Endocrinology)]
Dr. Chander M Malhothra [MBBS, MS, MCh]
Dr. Chander Shekar [MBBS, MS]
Dr. Chandra Bose
Dr. Chandragouda Dodagoudar [MBBS, MD - General Medicine, DNB - Medical Oncology]
Dr. Chandrakant Athalye [BDS, MDS]
Dr. Chandrakant Chavan [MD Medicine, DNB Cardiology, FACC, FESC]
Dr. Chandramani Panjabi
Dr. Chandrashekhar Kulkarni
Dr. Chandrashekhar N. Makhale [MD Medicine, DNB Cardiology, FACC, FESC]
Dr. Chandreyee Bhattacharya
Dr. Charan Reddy [MBBS, MD - General Medicine, DNB]
Dr. Charita Pradhan [MBBS, MS - General Surgery, DNB]
Dr. Charu Garg
Dr. Charu gauba
Dr. Charu Mithal
Dr. Charuchandra Joshi [MBBS, MD (Obstetrics & Gynaecology)]
Dr. Charudatt Vaity
Dr. Charudatta Chaudhari [MBBS, MS - General Surgery, DNB - Plastic Surgery]
Dr. Charudutt Apte [MBBS, MS - Neurosurgery]
Dr. Charusheela Sabane [MBBS, MD (Gynaecology)]
Dr. Charusheela Sabane [MBBS, MS - Obstetrics & Gynaecology]
Dr. Chetan K Sharma
Dr. Chetan Sharma [BAMS, MSc - Healthcare Management, NDDY]
Dr. Chhavi Agrawal
Dr. Chinmayee Agrawal [MBBS, MD, DM - Medical Oncology]
Dr. Chintan Gujrathi [MS, MCh Plastic & Reconstructive Surgery, DNB]
Dr. Chintan Mehta
Dr. Chirag Matravadia [MBBS, MD - Internal Medicine]
Dr. Chitra Sankar [MBBS, DCH (London), MRCP]
Dr. Clement Joseph
Dr. Col Akhil Mishra V S M [MBBS, MD, DM]
Dr. Col V Hariharan [MBBS, MD(Medicine), DM (Cardiology- AIIMS), FICA(USA) FISE FICC]
Dr. CS Narayanan [MBBS, MD - Medicine, DM - Neurology]
Dr. D B Usha Rajinikanthan [MBBS, DGO, MD - Obstetrics & Gynaecology]
Dr. D K Agarwal [MBBS, MD, DM (Neph), DNB (Neph), MNAMS(Neph), MAMS(Neph), FICP, FISN]
Dr. D K Bhargava [MD, PHD, FACG, AGAF]
Dr. D M Mahajan [MBBS, MD - Dermatology and Venerology]
Dr. D. Sureshkumar
Dr. D.V Singh
Dr. Daksh Chandra
Dr. Damanjit Duggal [MD Tuberculosis & Chest Diseases, IDCCM]
Dr. Danish Hasan Kazmi
Dr. Darshan B S
Dr. Darshan K. Patel
Dr. Darshan Krishnappa
Dr. Darshan Krishnappa
Dr. Darshan M. Patel [MDS Oral & Maxillofacial Surgery, FIHNOS Clinical]
Dr. Darshan Nimavat [MBBS, DTCD, DNB - Pulmonology, Fellowship in Pulmonology]
Dr. Darshana Rane
Dr. Davidson Devasia [MMBS, MD (Pediatrics), DM (Neurology), Fellowship in Paediatric Neurology and Epilepsy, Diploma (Pediatric Sleep Medicine)]
Dr. Davinder Kundra [MBBS, Diploma - Tuberculosis and Chest Diseases, DNB - Respiratory Diseases]
Dr. Davinder Singh Chadha
Dr. Debabrata Chakraborty
Dr. Debabrata Mukherjee
Dr. Debanjan Banerjee
Dr. Debapriya Mondal
Dr. Debarshi Chatterjee
Dr. Debashis Ghosh
Dr. Debashis Roy
Dr. Debasish Banerjee
Dr. Debasish Mitra
Dr. Debmalya Gangopadhyay
Dr. Debnath Dwaipayan [MBBS, MS(Gen. Surgery), DrNB (Neurosurgery)]
Dr. Debopam Chatterjee
Dr. Deep Goel
Dr. Deepa Divekar [MBBS, MD (Paediatrics)]
Dr. Deepa Giri [MBBS, DNB - Obstetrics & Gynaecology]
Dr. Deepa Maheshwari [Gold Medal In MBBS, MD - Obstetrics & Gynaecology ,Infertility Specialist]
Dr. Deepa N A [MBBS, DM - Neurology]
Dr. Deepa Sharma [MBBS, DCH, DNB]
Dr. Deepak Ahuja [MBBS, MD - Paediatrics]
Dr. Deepak Charles [MBBS, DM - Haematologist, BMT)
Dr. Deepak Chaudhary
Dr. Deepak Dubey [MBBS, MS - General Surgery, MCh - Urology, DNB - Urology, FRCS]
Dr. Deepak Govil
Dr. Deepak Gupta
Dr. Deepak Jayaprakash Kaddu [MBBS, MCh, MS, DNB - Urology]
Dr. Deepak Jha [MBBS, MS - General Surgery, Specialist Breast Surgery]
Dr. Deepak Kapoor
Dr. Deepak Keshav Bhangale
Dr. Deepak Kumar
Dr. Deepak Kumar Gupta [MBBS, MD - General Medicine, DM - Gastroenterology]
Dr. Deepak Kumar Mittal
Dr. Deepak P. Patkar [MBBS, DMRD, MD - Radiology]
Dr. Deepak P. Vyas
Dr. Deepak Padmanabhan
Dr. Deepak Raina [MBBS,MS-Orthopaedic Surgery]
Dr. Deepak Rosha [MBBS, MD]
Dr. Deepak Sarin
Dr. Deepak Sharma [MBBS, MS - General Surgery, MCh - Urology]
Dr. Deepak Subramanian [MS - General Surgery, MBBS]
Dr. Deepakala N [MBBS, MD - Paediatrics]
Dr. Deepakala N [MBBS, MD - Paediatrics]
Dr. Deepankar Bhattacharya [MD Pediatrics, MBBS, FNB Hemato-oncology and BMT]
Dr. Deepika Gumber [MBBS, DMRD - Radiology]
Dr. Deepika Parwan [MD - Pathology]
Dr. Deleep Kumar Gudipudi [MBBS, DNB - Radiotherapy]
Dr. Deshbandhu [MBBS, DNB - General Medicine]
Dr. Devasheesh Kamra [MBBS (MAMC) | MCh Neurosurgery AIIMS Delhi, Neurointerventional Surgery]
Dr. Devashish Tripathi [MBBS, PLAB, MRCP (UK)- General Medicine, FRCR (Oncology), Certificate of Completion of Training (CCT)- Clinical Oncology]
Dr. Devavrat Arya
Dr. Devayani Barve-Venkat
Dr. Devendra Chaukar
Dr. Devendra Singh Solanki [MBBS, MS - Orthopaedics]
Dr. Devendra Venkatramani [MS Ophthalmology]
Dr. Devendra Yadav [MBBS, MS - Orthopaedics]
Dr. Devi Prasad Shetty [renowned cardiac surgeon]
Dr. Devpriya Mitra [MBBS, MS, DrNB - Urology, FEBU]
Dr. Devyani Mahajan
Dr. Dhairyasheel Kanase [MBBS, MS (General Surgery), MCh (CVTS)]
Dr. Dhanalakshmi M
Dr. Dhananjay Kumar
Dr. Dhananjay Kumar [MBBS, MS - ENT Surgery]
Dr. Dhananjay Malankar
Dr. Dhananjaya Bhat
Dr. Dhanya Dharmapalan [MBBS, MD - Paediatrics, PG Diploma - Paediatric Infectious Diseases]
Dr. Dharam Chandrani
Dr. Dharam Pani Pandey
Dr. Dharemendra Singh
Dr. Dharitri Samantaray
Dr. Dharma Choudhary
Dr. Dharmaraj Yadav [MBBS, MS - Orthopaedics, Diploma - Orthopaedics]
Dr. Dharmendra Kumar [MBBS, MD - General Medicine]
Dr. Dharmendra Verma [MBBS, DNB - Ophthalmology]
Dr. Dharmesh Solanki
Dr. Dhawal Sharma [MBBS, MS, DNB - Neurosurgery]
Dr. Dheeraj Batheja [MBBS, MS - Orthopaedics, Specialty : Orthopaedics, Spine Surgery]
Dr. Dheeraj Gupta [MBBS, MS - Ophthalmology]
Dr. Dheeraj Kapoor [MBBS, MD - General Medicine, DM - Endocrinology]
Dr. Dheeraj Kumar Gandotra
Dr. Dheerja Babbar
Dr. Dhiren Shah
Dr. Dhiyanesh Krishnamoorthy [MBBS, MS - Ortho]
Dr. Dhrumin Sangoi
Dr. Dhruv Chaturvedi [MBBS, MS-General Surgery, M.Ch- Neuro surgery]
Dr. Dhwanee Thakkar [MD Paediatrics (Gold Medal, Pediatric Hematologic-Oncologist)
Dr. Digvijay Sharma
Dr. Diksha Goyal [MBBS, MD - Internal Medicine]
Dr. Dilip Bhalla [DNB - Nephrology, DM - Nephrology, MBBS, MD - Medicine]
Dr. Dilip Vyas [MBBS, MD - General Medicine]
Dr. Dilpreet Bajwa [MBBS, MS - ENT]
Dr. Dimple K Ahluwalia
Dr. Dinesh Arora [MBBS, MD - Transfusion Medicine]
Dr. Dinesh B Padole
Dr. Dinesh Bansal
Dr. Dinesh Kansal
Dr. Dinesh Katiyar
Dr. Dinesh Khullar
Dr. Dinesh Kumar Mehta
Dr. Dinesh Kumar Yadav
Dr. Dinesh Pendharkar
Dr. Dinesh Ramaswamy
Dr. Dinesh Sareen
Dr. Dinesh Singhal [MBBS, MS - General Surgery, DNB - Surgical Gastroenterology]
Dr. Dinesh Talwar
Dr. Dinesh Zirpe [MBBS, MS (General Surgery), DNB (General Surgery), DNB (Surgical Gastroenterology) - Gold Medalist]
Dr. Dipalee Borade [MBBS, Diploma - Radiology Therapy, DNB - Radiation Oncology]
Dr. Dipanjan Panda [MBBS, MD, DM]
Dr. Dipanjana Datta
Dr. Disha Saxena [MBBS, MD, DM - Nephrology]
Dr. Disha Saxena [MBBS, MD, DM - Nephrology]
Dr. Divya Bajpai
Dr. Divya Bansal [MBBS, DNB Pediatrics, DM - Clinical Haematology ]
Dr. Divya Chowdhry
Dr. Divya Gupta [MBBS, MD - Radiation Oncology]
Dr. Divya Gupta [MBBS, MD, DNB - Dermatology]
Dr. Divya Sehra [MBBS, MS - Obstetrics and Gynecology, DNB - Obstetrics and Gynecology]
Dr. Dodul Mondal
Dr. Durgatosh Pandey
Dr. Durva Kurkure
Dr. E A Padma Kumar
Dr. Easakiselvan Kamraj
Dr. Edul Nazir [MD Medicine]
Dr. Ehtesham [MBBS (Gold Medalist), CTCCM]
Dr. Ekta Soni Puri [MA IN APPLIED PSYCHOLOGY &PG DIPLOMA IN CLINICAL PSYCHOLOGY]
Dr. Erika Patel [MBBS, MD - Obstetrics & Gynaecology]
Dr. Esha Kaul [MBBS, Fellowship - Hematology]
Dr. EV Raman [MBBS, DLO, MS]
Dr. Evie Satheesan [BDS, MDS, Fellowship - Advanced Endodontics]
Dr. Faisal Mumtaz
Dr. Faizan Rahmani
Dr. Farah Atul Ingale
Dr. Faryad Husain [MBBS, IDCCM, CTCCM]
Dr. Farzana Batt [MBBS,FRCP]
Dr. Feroz Amir Zafar
Dr. Feroz Pasha [MBBS, MS]
Dr. G K Prakash [MBBS, MD, DNB - Nephrology]
Dr. G Karthikeyan [MBBS, MD - Radiology, Fellowship - Interventional Radiology]
Dr. G Karthikeyan [MBBS, MD - Radiology, Fellowship - Interventional Radiology]
Dr. G N Prasad
Dr. G S Nagaraja Prabhakar [MD - Anaesthesiology]
Dr. G. Venkatesh Kumar
Dr. G.K. Jadhav
Dr. Gagan Gautam
Dr. Gajanan Kanitkar [MS Surgery]
Dr. Gajanand Yadav [MBBS, MS - Orthopaedics]
Dr. Gajinder Kumar Goyal
Dr. Ganesh Jadhav [MBBS, MD, DNB]
Dr. Ganesh Kumar Mani [MBBS, MS in General Surgery, MCh -Thoracic Surgery]
Dr. Ganesh Nagarajan
Dr. Ganesh S Deogaonkar
Dr. Gangesh Gunjan [MBBS, MS, MCh - Neurosurgery]
Dr. Garima Garg Seth [MBBS, MD - Paediatrics, Fellowship in Allergy & Immunology]
Dr. Garima Nirmal [MBBS, MD, DM]
Dr. Garima Singh [MBBS, MD - Radiotherapy]
Dr. Garvit Chitkara
Dr. Gaurav Agrawal [FNB (Pediatric Cardiology), MD (Pediatrics), MBBS, MIAP]
Dr. Gaurav Batthla [MBBS, DNB - Anaesthesia, IDCCM]
Dr. Gaurav Chanana
Dr. Gaurav Chaubal
Dr. Gaurav Dixit [MBBS, MD - Medicine, DM - Clinical Haematology]
Dr. Gaurav Ganeshwala [MD Medicine, DM Cardiology]
Dr. Gaurav Goel
Dr. Gaurav Jain [BDS, MDS]
Dr. Gaurav Kharya [Pediatric Hematologic-Oncologist]
Dr. Gaurav Kumar [MBBS, MD Pediatrics, DNB Pediatrics]
Dr. Gaurav Kumar [MBBS, MS - General Surgery, DNB - Cardiothoracic Surgery]
Dr. Gaurav Patil
Dr. Gaurav Pawale [MBBS, DGO, FMAS, PGDHHM]
Dr. Gaurav Rastogi [MBBS, MS - Orthopaedics]
Dr. Gaurav Sagar [MBBS, MD, DNB]
Dr. Gaurav Sapra [MBBS, MS - ENT]
Dr. Gaurav Sapra [MBBS, MS - ENT]
Dr. Gaurav Shrivastav [MBBS, MS (General Surgery), FMAS, FIAGES, FARIS (Robotic)]
Dr. Gaurav Tyagi [MBBS, MS, MCh (NIMHANS) Fellowship Skull Base Surgery, Osaka, Japan Former Faculty Department of Neurosurgery, NIMHANS, Bengaluru]
Dr. Gauri Barve [MBBS, MD (Gynaecology)]
Dr. Gauri Belsare [MS ENT]
Dr. Gautam Banga [MBBS, MS - General Surgery, MCh - Urology]
Dr. Gautam Dhar Choudhury
Dr. Gautam JK. Tawari
Dr. Gayatri Deshpande
Dr. Gayatri Sasikumar [MBBS, DA, DNB - Anaesthesiology]
Dr. Gazal Garg
Dr. Geet Bajpai
Dr. Geeta Chadha [MBBS, MD, FOGSI (Obs & Gyan)]
Dr. Geeta Kathuria
Dr. Geetha S [MBBS, DOMS, DNB]
Dr. Geetu Gaba [MBBS, Diploma - Gynecology and Obstetrics, DNB - Obstetrics and Gynecology]
Dr. George Karimundackal
Dr. Ghazal Tansir [DM (Medical Oncology), MD (Internal Medicine), DNB (Internal Medicine), MBBS]
Dr. Girija Suresh
Dr. Giriraj Bora [Liver Transplantation and GI and HPB Surgery]
Dr. Girish Date [MBBS, MD (General Medicine)]
Dr. Girish Date [MBBS, MD - Medicine]
Dr. Girish Kakade [MBBS, MD (Medicine), DNB (Rheumatology)]
Dr. Girish Nair [MBBS, MD, DM - Neurology]
Dr. Girish Parmar
Dr. Girish Raheja [MBBS, MS]
Dr. Girish Rai [MBBS (AIIMS Delhi), MS (AIIMS Delhi) Otolaryngology]
Dr. GIRISH SABNIS
Dr. Gitanjali Kochar [MBBS, MRCP(UK)]
Dr. Govind Eriat [MBBS, DNB, Fellowship - Leukemia and Bone Marrow Transplantation]
Dr. Gulshan Rohra
Dr. Gulzar Ali [MBBS, MD - Pathology]
Dr. Gunjan Budhiraja [MBBS, Diploma - Ophthalmology, Cataract – Cornea – Lasik/Specs Removal]
Dr. Gunjan Verma [MBBS, MD - Dermatology]
Dr. Gurinder Bedi
Dr. Gurneetsingh Sawhney
Dr. Gurpratap Singh Grewal
Dr. Gurtejsingh Sardar [MD Radio Diagnosis]
Dr. Guruprasad S Bhat [MBBS, MD, MRCP (UK), FRCP (Edinburgh)]
Dr. Gurvinder S. Sawhney
Dr. Gyanam Misra [MBBS, MD - Paediatrics]
Dr. Gyanti R.B. Singh [MBBS, MD, DNB - Cardiology]
Dr. H Sudarshan Ballal [MD, FRCP (London), Board Certified Internal Medicine, Nephrology, Critical Care (USA)]
Dr. H. S. Bhatyal
Dr. H.N Bajaj
Dr. H.S. Chhabra
Dr. Hamza Shaikh [MBBS, Diploma in Orthopaedics, DNB - Orthopedic Surgery]
Dr. Hanna Angel [Meleth [MBBS, MD-Pediatrics, DrNB -Neurology]
Dr. Har Prakash Garg [MBBS, MS]
Dr. Hardeep Singh [MBBS, MS, MCh Plastic Surgery]
Dr. Hardik Dhamsania [MBBS, MS Orthopedics]
Dr. Hardik Shah
Dr. HARESH DODEJA
Dr. HARESH MANGALANI
Dr. Hari Goyal [MBBS, MD - General Medicine, DM - Medical Oncology]
Dr. Hari Priya A
Dr. Harin Kamleshbhai Vyas
Dr. Harish Kumar
Dr. Harish Mohanty
Dr. Harish Nayak [MBBS, DOMS, MS (Bombay Univ), CCT Ophthalmology (UK), FRCS (Glasgow), FRCOphth (London)]
Dr. Harish Pathak
Dr. Harish Saini
Dr. Harish Verma [Surgical Oncology]
Dr. Harit Kumar Chaturvedi [M.B.B.S, M.S. (General Surgery), M.Ch. (Surgical Oncology)]
Dr. Harmeet Kaur Sodhi [MBBS, MD - Radiology]
Dr. Harmeet Malhotra [MBBS, MD, DGO, FICOG]
Dr. Harmeet Singh Kapoor
Dr. Harnarayan Singh [MBBS, MS - General Surgery, M.Ch - Neurosurgery]
Dr. Harpreet Singh [MBBS, MS - ENT]
Dr. Harsh Dua [MBBS, MD]
Dr. Harsh Kumar
Dr. Harsh Sapra
Dr. Harsha G N
Dr. Harsha Jain [MBBS, MD (Pulmonary Medicine), DM (Pulmonary, Critical Care & Sleep Medicine)]
Dr. Harsha Jauhari [MBBS, MS - General Surgery, Renal Transplant Specialists]
Dr. Harsha Kumar
Dr. Harshad Limaye
Dr. Harshad Nikte [MBBS, MS - ENT, DNB - ENT]
Dr. Harshad Parekh
Dr. Harshal Ekatpure [MBBS, MD Internal Medicine, DNB Endocrinology]
Dr. Harshavardhan Hegde
Dr. Harshit Chakshota
Dr. Harshit Garg [MBBS, M.S(General Surgery), M.Ch (Urology)]
Dr. Harshita Tripathi
Dr. Hasmukh Gujar [MBBS, DNB - Cardiology]
Dr. Hasmukh Gujar [MBBS, MD - General Medicine, DM - Cardiology]
Dr. Hasmukh Ravat
Dr. Havind Tandon [MBBS, MS]
Dr. Haytham Mutasim Taha [MBBS, Certified in Internal Medicine]
Dr. Heenaa Mackasare
Dr. Hemalata Arora
Dr. Hemanandini Jayaraman [MBBS, MD, DGO, FCPS, DNB, MRCOG]
Dr. Hemant B. Tongaonkar
Dr. Hemant K Gogia [MBBS, MD - Pediatrics]
Dr. Hemant K Kalyan [MBBS, MS (Orthopaedics) Univ of Bombay, D Ortho, FCPS, D Sports Med]
Dr. Hemant Patankar [MBBS, MS - Orthopaedics]
Dr. Hemant Patil
Dr. Hemant Sant [MBBS, DNB - General Medicine, DM - Neurology]
Dr. Hemant Sant [MBBS, MD (Internal Medicine), DM (Neurology)]
Dr. Hemant Sharma [MBBS, DNB - Orthopedics Surgery, Diploma in Orthopaedics, Robotic Surgery]
Dr. Hemanth G N [MBBS, MS, MCh]
Dr. Hemanth Kumar
Dr. Hemanth Prajwal M [MBBS, MS - General Surgery, DrNB - Peripheral Vascular Surgery]
Dr. Hiba Siddiqui
Dr. Himani Sharma [MBBS, DNB - Obstetrics & Gynaecology]
Dr. Himanshi Joshi
Dr. Himanshu Batra [MBBS, Diploma - Child Health, DNB - Pediatrics]
Dr. Himanshu Bhargava [MS orthopaedics]
Dr. Himanshu Champaneri [MBBS, MS, DNB-Neurosurgery]
Dr. Himanshu Dewan [MBBS, DNB, Fellowship - Critical Care Medicine]
Dr. Himanshu Khanna
Dr. Himanshu Koyani [MBBS, MS General Surgery, MCh]
Dr. Himanshu Pratap
Dr. Himanshu R. Mehta [MBBS, MS - Opthalmology, Fellowship - Vitreo Retinal Surgery]
Dr. Himanshu Sharma [MBBS, MD, DM - Endocrinology]
Dr. Himanshu Shukla
Dr. Himanshu Tyagi
Dr. Himesh Gandhi [MS General Surgery, MCh Urology]
Dr. Hirak Mazumder
Dr. Hiren N. Doshi
Dr. Hitendra K Garg [MBBS,MD,DM]
Dr. Hitesh Garg [MS - Orthopaedics, MBBS Spine Surgery & Scoliosis Surgery]
Dr. Hitesh Hans Baweja [BDS, MDS - Oral and Maxillofacial Surgery]
Dr. Hitesh Kumar
Dr. Hitesh Pant [MBBS, Diploma in Child Health]
Dr. Hiteshi Dhami Shah
Dr. Hompriya Issar [MBBS, DNB - Surgery, DNB - Neurosurgery]
Dr. HRISHIKESH P SALGAONKAR
Dr. HRISHIKESH TADWALKAR
Dr. Huma Noor [MBBS, MD - General Medicine, DM - Medical Oncology]
Dr. Hunaid Hatimi
Dr. I P S Kochar [MBBS, MD, MAMS, MRCPH]
Dr. Ila Gupta [MBBS, MS - Obstetrics & Gynaecology]
Dr. Imran Khan [MBBS, MD, DNB - Medical Oncology]
Dr. Indoo Ammbulkar
Dr. Indradip Maity
Dr. Indrajit Agrawal [MBBS, MD - Internal Medicine, DM - Rheumatology]
Dr. Indranil Ghosh
Dr. Indrapati Singh
Dr. Indu Khosla
Dr. IPS Oberoi [MBBS, MS - Orthopaedics, Robotic Joint Replacement & Arthroscopy Surgery]
Dr. Irfana Mohammed [MBBS, DNB-Internal Medicine,Fellowship Infectious diseases]
Dr. IS Mehta [BDS]
Dr. Ishan Gupta [MBBS, DNB RESPIRATORY DISEASES]
Dr. Ishrat Majid Mir
Dr. Ishu Gupta [MBBS, MD - General Medicine, DM - Oncology]
Dr. Ishwar Bohra[Robotic Joint replacement & Orthopedic Surgeon]
Dr. Ishwar Zanwar [MBBS, DNB (General Medicine), DNB (Cardiology), FESC, FSCAI]
Dr. Itisha Chaudhary[MBBS, DNB - General Surgery, MCh - Surgical Oncology]
Dr. J AL Ranganath
Dr. J Maheshwari
Dr. J S Kumar
Dr. J V Srinivas
Dr. J. S. Rajkumar
Dr. J.C Vij
Dr. Jagadish Shejul [MBBS, MD (Radiation Oncology)]
Dr. Jagat Jot Singh Gill [MBBS, MD, DM - Gastroenterology]
Dr. Jagdish Chander Suri
Dr. Jagdish Hiremath [MD Medicine, DM Cardiology]
Dr. Jagruti Sanghvi
Dr. Jai Parkash Gurawalia
Dr. Jai Ranjan Ram
Dr. Jaichitra Suresh
Dr. Jaideep Gambhir [MBBS, MD - Psychiatry]
Dr. Jaidrath Kumar
Dr. Jaini Lodha Bhandari
Dr. Jaising Shinde [MBBS, MS - General Surgery, Fellowship - Laparoscopic Surgery]
Dr. Jaising Shinde [MS (General Surgery), FICS, FACRSI]
Dr. Jaisom Chopra [MBBS, MS - General Surgery, Vascular Surgeon]
Dr. Jasvinder Paintal [MBBS, Diplomate In Internal Medcine]
Dr. Jatin Ahuja
Dr. Jatin Kothari
Dr. Jawaharlal Goyal [ DNB - Ophthalmology, MD, MBBS ]
Dr. Jay Relan [MBBS, MD, DM - Paediatric Cardiology]
Dr. Jaya Kumar Agarwal [MBBS , DGO , DNB (obstetric and gynecology) DGE diploma in Gyne endoscopy (Germany )]
Dr. Jayanivash J [MBBS, MD - General Medicine, DM - Nephrology]
Dr. Jayant Barve
Dr. Jayant Jaswal [MBBS, MS - ENT, DNB - ENT and Head and Neck Surgery]
Dr. Jayant Kumar Hota [MBBS, MD, DM (Nephrology)]
Dr. Jayant Shah [MS Orthopaedics]
Dr. Jayanta Kumar Gupta
Dr. Jayanta Sharma
Dr. Jayanth Reddy
Dr. Jayapriya Ramas
Dr. Jayasurya R
Dr. Jaydeep Desai [MBBS, MD (General Medicine), DNB (Cardiology) Gold Medalist]
Dr. Jaydev Panchawagh [MBBS, MS - General Surgery, MCh - Neurosurgery]
Dr. Jaydev Panchawagh [MS (General Surgery), M.Ch (Neurosurgery)]
Dr. Jaydip Bhadra Ray
Dr. Jayesh Sardhara
Dr. Jem Kalathil [MBBS,MS Onco Surgery,FRCS]
Dr. Jesal Sheth
Dr. Jheel Ambike [MD, DNB Dermatology]
Dr. Jibak Bhattacharya
Dr. Jigarsinh Jadeja [MBBS, MS, MCh - Neurosurgery (PGI Chandigarh)]
Dr. Jigna Ganatra [MBBS, DGO, Diploma in Gynae Endoscopy, CIMP France]
Dr. Jignesh Ashwinbhai Gandhi
Dr. Jignesh Gandhi
Dr. Jimit Choudhary
Dr. Jithin Jagan Sebastian [MBBS, MS - General Surgery, MCh - Vascular Surgery]
Dr. Josna Sabnis [MBBS, MS, DNB (Neurosurgery)]
Dr. Joy Debasis Ghose [MBBS, MS - General Surgery, MCh - Surgical Oncology]
Dr. Joy Varghese
Dr. Joydeep Ghosh
Dr. Juhi Agrawal [MBBS, MS - General Surgery, MCh - Burns, Plastic and Reconstructive Surgery]
Dr. Juili Kulkarni
Dr. Jumana Yusuf Haji
Dr. Jyoti Bala Sharma [MBBS, MD - General Medicine, DM - Neurology]
Dr. Jyotika Jain
Dr. Jyotirmaya Sahoo [MBBS, MD, DM - Cardiology]
Dr. K Hemanth Kumar [MBBS (BMCRI), MS (PGIMER), DNB, MCh Surgical Gastroenterology (PGIMER)]
Dr. K Jayanthi
Dr. K K Pandey [MBBS, MS, Ph.D (CTVS)]
Dr. K K Saxena [MBBS, MD, DM]
Dr. K M K Varma [MBBS, MS, FRCS, MCh]
Dr. K N K Shetty [MBBS, MRCP, Fellow in Gastroenterology (London)]
Dr. K R Suresh Bapu [MBBS, MS - General Surgery, MCh - Neuro Surgery]
Dr. K R Vasudevan
Dr. K S Iyer
Dr. K Sharmatha [MBBS, DDVL - Diploma in Dermatology, Venereology & Leprosy]
Dr. K Sridhar
Dr. K Subramanyan
Dr. K Thiruppathi
Dr. K VengadaKrishnan [MBBS, MD - General Medicine]
Dr. K VengadaKrishnan [MBBS, MD - General Medicine]
Dr. K. Balamurugan
Dr. K. Jai Shankar
Dr. K. Matchavel
Dr. K. S. Rana [MBBS, MD - Pediatrics]
Dr. K. Vasanth
Dr. K.R Balakrishnan
Dr. Kadam Nagpal [MBBS, MD - Medicine, DM - Neurology]
Dr. KAILASH KOTHARI
Dr. Kailash Nath Singh [MBBS, MD , DNB]
Dr. Kajal Das
Dr. Kalaivanan Kanniyan
Dr. Kalpana Nagpal [MBBS, MS - ENT, DNB - Otorhinolaryngology]
Dr. Kalpana R
Dr. Kalpana Sarangi
Dr. Kalyan Dalave [MBBS, DNB Dermatology]
Dr. Kalyanpury Jawaharlal Choudhury [M.B.B.S ,M.D. (Anaesthesia),M.N.A.M.S F.W.A.C.S]
Dr. Kamal Ahmad [MBBS, MD (Medicine)]
Dr. Kamal Kiran
Dr. Kamal Verma [MBBS, MD - Internal Medicine]
Dr. Kamal Verma [MBBS, MS - General Surgery, MCh - Neurosurgery]
Dr. Kamal Verma[MBBS, Diploma in Radio Therapy, DNB - Medical Oncology]
Dr. KAMALJIT SINGH
Dr. Kameshwar Prasad [MBBS, MD General Medicine , FAMS, DM Neurology]
Dr. Kamran Ali [MBBS, DNB - General Surgery]
Dr. Kanchan S. Gadkari
Dr. Kanchana Pillai
Dr. Kanchankumar Bhagyawant [MD Paediatrics, PGPN Boston]
Dr. Kanika Batra Modi [MBBS, DNB - Obstetrics & Gynecology]
Dr. Kanika Gupta
Dr. Kanika Narang [Msc. In Dietetics and Food Service management from Institute of Hotel Management, Catering and Nutrition. Bsc. In Home science from Institute of Home Economics, Delhi University]
Dr. Kannan Subramanian [DNB (General Medicine), DM (Haematology)]
Dr. Kant Jogani [MBBS, MS - General Surgery, MCh - Neurosurgery]
Dr. Kant Shah
Dr. Kanu Verma
Dr. Kapil Agarwal [MBBS, MD - General Medicine, DM - Neurology]
Dr. Kapil Borawake [MBBS, DNB (General Medicine)]
Dr. Kapil Goyal
Dr. Kapil Gupta [MBBS, DNB - Emergency Medicine, Fellowship]
Dr. Kapil Jamwal
Dr. Kapil Kochhar [MBBS, MS - General Surgery]
Dr. Kapil Kumar [MBBS, MS(Ortho), MCh(Orth), FRCS, FRCS(Orth)]
Dr. Kapil Mago [BPT, MPT - Physiotherapy & Rehabilitation]
Dr. Kapil Virpariya
Dr. Karan Annapur Kumarswamy [MBBS, DNB, FVRS]
Dr. Karan Deshmukh [MBBS, MD - Respiratory Medicine]
Dr. Karuna Ratwani
Dr. Karunesh Kumar [MBBS, MD (Pediatrics), FNB (PGHN), RCPCH Fellowship (London, UK)]
Dr. Kashif Hafeez [MBBS, FRCP, MRCP, Endocrinologist & Diabetologists]
Dr. Kashinath Shridhar Bangar [DNB Anaesthesia]
Dr. Kashish Kalra [MD - Dermatology , Venereology & Leprosy, MBBS]
Dr. Kaurabhi Zade [MBBS, Dip (Radio Diagnosis), DNB, EDSI, EBIR]
Dr. Kaushal Kant Mishra
Dr. Kaushal Kejriwal [MBBS, MS - General Surgery, FIAGES]
Dr. Kaushal Kishore Yadav [MBBS, MS General Surgery, MCh Surgical Oncology]
Dr. Kaushal Malhan
Dr. Kaushic A [MBBS, MD, DM - Neuroanaesthesia]
Dr. Kaushik Bhojani
Dr. Kaushik Nandy
Dr. Kaushik Sheth [MD Medicine, DM Cardiology]
Dr. Kaustabh Choudhuri
Dr. Kaustav Talapatra
Dr. Kaustubh Das
Dr. Kaveshwar Ghura
Dr. Kavitha Vinayak [MBBS, MS - OBG, Fellowship in Fetomaternal Medicine]
Dr. Kavya Mallikarjun [MBBS (JSS Mysore), MD Paediatrics (JSS Mysore), DNB Paediatric Cardiology (Narayana Hrudayalaya)]
Dr. KD Soni [MBBS, MS - Orthopaedics, MCh - Orthopaedics]
Dr. Kedar Kulkarni [DNB (Internal Medicine), DNB (Cardiology)]
Dr. Kedar Patil [DNB General Surgery, FNB Minimal Access Surgery]
Dr. Kedar Tilwe
Dr. Keni Ravish Rajiv
Dr. Keshav Naithani [BDS, MDS - Oral & Maxillofacial Surgery]
Dr. Keshava R
Dr. Ketan Pai [MBBS, DNB (General Surgery), DNB (Urology)]
Dr. Ketan Pai [MBBS, FCPS - General Surgery, DNB - General Surgery, DNB - Urology/Genito-Urinary Surgery]
Dr. KETAN PANDITPAUTRA
Dr. Ketan Vartak [MS, MCh, DNB (Urology)]
Dr. Ketul Shah [MBBS, MS - General Surgery, M.Ch - Surgical Gastroenterology]
Dr. Khushbu Goel
Dr. Kinsuk Das
Dr. Kiran Arora [MBBS, MD - Obstetrics & Gynaecology, specialist IVF, Reproductive Medicine]
Dr. Kiran Gadre [MDS]
Dr. Kiran Kharat [MS Ortho, MSc Ortho London, LAMP]
Dr. Kiranbala Dash [MD (Obst & Gyn), FMAS]
Dr. Kiranmoy Sarangi
Dr. Kirtee Mishra [MBBS, MRCP (UK), IDCCM]
Dr. Kirti Chandrashekhar Sabnis
Dr. Kishalaya Karan
Dr. Kishan Panicker G [BDS, MDS, MOI (USA)]
Dr. KISHORI KADAM
Dr. Koteshwara Prasad
Dr. kranjit singh Narang
Dr. Krantikumar Rathod [MBBS, MD - Radio Diagnosis]
Dr. Krishna Kumari
Dr. Krishna Shama Rao [BDS, MDS - Oral & Maxillofacial Surgery, MBBS, FRCS - Oral and Maxillofacial Surgery, FDSRCS - Fellowship in Dental Surgery]
Dr. Krishna Shankar Singh
Dr. Krishna Subramony Iyer
Dr. Krishnamohan Yarlagadda [Robotic, Laparoscopic, Bariatric & GI Surgeon]
Dr. Kriti Menon [B.Tech (Genetic Engineering), PGDPC, BGCI Level II Certified Genetic Counselor]
Dr. Krupa Thakkar
Dr. KS Brar [MBBS, MD - General Medicine, DNB - General Medicine, LTT Endocrinology, AIIMS ]
Dr. Kshitij Raghuwanshi [MS General Surgery, MCh Urology]
Dr. Kshitij Thoke [MBBS, MD, Diploma - Orthopaedics]
Dr. Kshitiz Sharan [MBBS, MD, DM - Gastroenterology]
Dr. KSS Bhat
Dr. Kulbhushan Attri [MBBS, MS, D-Orth, MCh (Liverpool, UK)]
Dr. Kulbhushan Singh Dagar
Dr. Kuldeep Arora [MBBS, MD - General Medicine, DM - Cardiology , Interventional Cardiologist]
Dr. Kuldeep Dole [MBBS, MS (Ophthalmology)]
Dr. Kuldeep Malik
Dr. Kuldeep Singh [MBBS, MS - General Surgery, MCh - Plastic Surgery]
Dr. Kumar Doshi
Dr. Kumar Narayanan
Dr. Kumar Rishikesh [MBBS, MD (Medicine), DNB (Medical Oncology)]
Dr. Kumar Satyakam
Dr. Kumar Shetty
Dr. Kumud Rai [MBBS, MS in General Surgery]
Dr. Kunal Banavali
Dr. Kunal Das [MBBS, MD - General Medicine, DM - Gastroenterology]
Dr. Kunal Kumar [MBBS, MD Psychiatry]
Dr. Kunal Nigam [MBBS, MS - ENT, Fellowship in Cochlear Implants]
Dr. Kunal Raj Gandhi [MD - General Medicine, DM - Nephrology, MBBS]
Dr. Kush Sharma [MBBS, MD - Anaesthesiology]
Dr. Kushal Bairoliya
Dr. Kushal Nag
Dr. L R Seth [MBBS, MS, DOMS]
Dr. Lakshmi Kant Tripathi [MBBS, MD - Internal Medicine, DM - Nephrology]
Dr. Lalit Kumar Lohia [MBBS, MS - Orthopaedics]
Dr. Lalit Kumar [MBBS, MD - Medicine, DM - Medical Oncology]
Dr. Lalit Kumar [MBBS, MS - Orthopaedics, Fellowship in Joint Replacement]
Dr. Lalit Rawal [MD Paediatrics]
Dr. Lalit Shimpi [MBBS, MD - General Medicine, DM - Gastroenterology, Fellowship - Pancreatobiliary Endoscopy & EUS]
Dr. Lalit Verma
Dr. Lalit Verma
Dr. Lalit Verma
Dr. Lalita Badhwar [MBBS, MD]
Dr. Lata Patil [MBBS, MD Medicine]
Dr. Latha Madhavan
Dr. Lav Kaushik [MBBS, MD - Psychiatry]
Dr. Leena N Sreedhar [MBBS, MD - Obstetrics and Gynecology, Fellowship]
Dr. Leena Sharma [BDS]
Dr. Leera Lobo
Dr. Lekh Ram Sharma [MBBS from Indira Gandhi Medical College, Shimla (HP University) 1977 MD (Medicine) from AFMC Pune, (Pune University) - 1982]
Dr. Lekha Pathak
Dr. LILADHAR CHANDAN
Dr. Litan Naha Biswas
Dr. Lokendra Kumar Yadav
Dr. Lokesh Garg [MBBS, MS - Orthopaedics, Fellowship in Orthopaedic Oncology]
Dr. Lokesh Kumar
Dr. Lovkesh Anand [MBBS, MD - General Medicine, DM - Hepatology]
Dr. M Gowrishankar [MBBS, MD - General Medicine]
Dr. M Gowrishankar [MBBS, MD - General Medicine]
Dr. M Madhavan
Dr. M N Sehar [MBBS, MS, Dip.Orth, FRCS, FRCS Orth (UK)]
Dr. M Padmaja Bhattacharya
Dr. M R C Naidu [MBBS, DNB - Neurosurgery, MCh - Neurosurgery]
Dr. M Ram Prabahar [MBBS, MD, DNB - General Medicine]
Dr. M S S Mukharjee
Dr. M Sudhakar Rao
Dr. M. Agila Saravanan
Dr. M. Elango
Dr. M. K. Ayyappan
Dr. M. K. Mutatkar [Diploma Radio Diagnosis]
Dr. M. Krishna Kumar
Dr. M. Roshan Kumar
Dr. M.A Mir [Gastroenterologist and Endoscopist ]
Dr. Madhav Balajirao Dawkore
Dr. Madhavi Dadwe [MBBS, MD General Medicine, DM Nephrology]
Dr. Madhu Roy [MBBS, MD]
Dr. Madhukar Bhardwaj
Dr. Madhulika Sinha
Dr. Madhur Bhatia [MBBS, MS - Ophthalmology]
Dr. Madhur Malik
Dr. Madhuri S Pattiwar [MBBS, MS - Ophthalmology]
Dr. Madhuri Singh
Dr. Magimairaj D J
Dr. Mahavir Modi [MD Tuberculosis]
Dr. Mahendra Kumar Sharma [MBBS, MS, DNB - Urology]
Dr. Mahendra Narwaley [M.Ch-CTVS, MS-General surgery, MBBS]
Dr. Mahesh Bhagwat [MBBS, MD (Radiology)]
Dr. Mahesh Chandra Garg [MBBS, MD ( Medicine ), MRCP (UK), FRCP(London), FRCP ( Glasgow) FRCP (Glasgow)]
Dr. Mahesh Chavan [MBBS, MD - General Medicine, DNB - Endocrinology]
Dr. Mahesh Goenka
Dr. Mahesh Kumar Gupta [MBBS, MD - Internal Medicine, DM - Gastroenterology]
Dr. Mahesh Wadhwani[MBBS, MS - General Surgery, MCh - Cardiothoracic and Vascular Surgery]
Dr. Mahin Nallasivam R R
Dr. Maitrey Joshi [MBBS, MS, MCh - Urology]
Dr. Maitri Chaudhuri
Dr. Malay Nandy [MBBS, MD - General Medicine, DM - Medical Oncology]
Dr. Mallikarjun Kalashetty [MBBS, MD - General Medicine, DM - Clinical Haematology]
Dr. Mallikarjuna Reddy N [MBBS, MS, MCh, DNB-Urology, Fellow European Board of Urology]
Dr. Mallinath Mukherjee
Dr. Malvika Srivastava [MBBS, MD - Pathology]
Dr. Mamatha Shetty
Dr. Man Mohan Mehnidratta
Dr. Manabendra Naskar
Dr. Manabendra Nath Basu Mallick
Dr. Manal Elredy [Gynecologist, Obstetrician and Urogynecology]
Dr. Manan Doshi [MD, DNB (Nephrology)]
Dr. Manas Kumar Panigrahi [MBBS, MCh - Neurosurgery, Fellowship - Skull Base Cerebrovascular Surgery]
Dr. Manas Mengar
Dr. Manash Biswas
Dr. Manash Saha
Dr. Manav Suryavanshi
Dr. Manav Wadhwan
Dr. Mangesh Udar [DNB (Internal Medicine), DNB (Neurology)]
Dr. Manik Sharma [MCh - Plastic Surgery, MBBS, MS - General Surgery]
Dr. Manimegalai
Dr. Manish Agarwal
Dr. Manish Baldia [MBBS, MCh Neurosurgery]
Dr. Manish Garg
Dr. Manish Itolikar
Dr. Manish Jain [MBBS, MD - Psychiatry ]
Dr. Manish Jain [MBBS, MS - General Surgery, DNB - Surgical Gastroenterology]
Dr. Manish Kumar Gaur[ MCh Surgical Oncology (AIIMS, New Delhi), MBBS, MS (General Surgery)]
Dr. Manish Kumar Jain
Dr. Manish Machave [MBBS, MD Obs & Gynae]
Dr. Manish Mahajan [MBBS, MD - General Medicine, DNB - Neurology]
Dr. Manish Nanda [MBBS, MS - General Surgery, MCh - Plastic Surgery]
Dr. Manish Pahuja [MBBS, DA - Anaesthesiology]
Dr. Manish Sabnis [MBBS, DNB - Neurosurgery]
Dr. Manish Salunkhe [MBBS, MD (General Medicine), DM (Neurology), Fellowships in Stroke and Neuro-interventions]
Dr. MANISH SHIRSAT
Dr. Manisha Chakrabarti [MBBS, MD Pediatrics, FNB - Pediatric Cardiology]
Dr. Manisha Dhobe [MD Medicine]
Dr. Manisha Mendiratta
Dr. Manjeeta Das [MBBS, MD - Internal Medicine]
Dr. Manju Aggarwal [MBBS, DNB - General Medicine, Fellowship in Nephrology, DNB - Nephrology]
Dr. Manmohan M. Kamat
Dr. Mannu Bhatia [DNB in Orthopedic Surgery, MBBS]
Dr. Manohar Joshi [MBBS, MD - General Medicine, Fellowship - Rheumatology]
Dr. Manohar Shaan
Dr. Manoj Bauskar [MBBS, DLO, DNB - ENT]
Dr. Manoj Bhise [MBBS, MD (General Medicine), DM (Cardiology)]
Dr. Manoj Bhise [MD - General Medicine, DM - Cardiology]
Dr. Manoj Durairaj [MBBS, MS - General Surgery, MCh - Cardio Thoracic & Vascular Surgery]
Dr. Manoj Durairaj [MS (General Surgery), M.Ch (Cardiac Surgery), FACC, D.Litt., Advanced Fellowship in Cardiac Surgery]
Dr. Manoj Jain
Dr. Manoj Kumar Khandelwal
Dr. Manoj Kumar Mahata
Dr. Manoj Kumar S [MBBS, Diploma in Orthopaedics, DNB - Orthopedics Surgery]
Dr. Manoj Miglani
Dr. Manoj Naik [MBBS, MD (General Medicine), DNB (General Medicine)]
Dr. Manoj Naik [MBBS, MS - Orthopaedics]
Dr. Manoj Padman
Dr. Manoj Shrivastav [MBBS, DNB General Surgery]
Dr. Manoj Yadav [MBBS, MD, DM - Gastroenterology]
Dr. Manoj Yadav [MBBS, MD, DM - Gastroenterology]
Dr. Mansi Munshi [MD Radiotherapy]
Dr. Manu Bora
Dr. Manvinder Singh Sachdev [MBBS, Diploma in Paediatrics, MD - Pediatrics, FNB - Pediatric Cardiology]
Dr. Masood Habib
Dr. Mayank Patil [BDS, MDS - Oral & Maxillofacial Surgery]
Dr. Mayur Mhatre [MBBS, MS - General Surgery, MCh - Neurosurgery]
Dr. Mayur Satish Shah
Dr. Meenakshi Dua
Dr. Meenal Patil [MD, DA]
Dr. Meenesh Juvekar
Dr. Meenu Agarwal [MBBS, MD Obs & Gynae]
Dr. Meenu Malik
Dr. Meenu Walia
Dr. Meera Luthra
Dr. Meet Kumar [MBBS, MD - Internal Medicine, DM - Clinical Hematology]
Dr. Megha Rustagi [MBBS, MD - Paediatric Gastroenterology]
Dr. Meghana Patwardhan
Dr. Meghna Chawla [MD Paediatrics, DNB Paediatrics]
Dr. Mercy Samuel [MBBS, MFM Family Medicine]
Dr. MG Pillai [MBBS, MD - General Medicine, DM - Cardiology]
Dr. Mihir Bapat
Dr. Mihir Raman Gangakhedkar
Dr. Mihir S. Raut [MBBS, DNB - Medicine, Post Doctoral Fellowship - Diabetology]
Dr. Milan Chetri
Dr. Milind Gadkari [MBBS, MD (General Medicine), MRCP, FRCP, FACC]
Dr. Milind Padmakar Hote
Dr. MILIND SAWANT
Dr. Milli Karia [BDS, MDS - Conservative Endodontics]
Dr. Minakshi Bansal [MBBS, MD PEDIATRICS, FIAP (PHO)]
Dr. Minakshi Fullara (PT)
Dr. Mini Nampoothiri [MBBS, DNB - Obstetrics and Gynecology, Diploma - Obstetrics and Gynaecology]
Dr. Mini Ravi [Gynecologist, Obstetrician & Reproductive Medicine specialist]
Dr. Minish Jain [MBBS, MD General Medicine, DM Medical Oncology]
Dr. Minoo Fazilat
Dr. Mir Reza Kamal
Dr. Mirza Masoom Abbas
Dr. Mithilesh Yadav [MBBS, MS, MCh - Urology]
Dr. Mithun C B
Dr. Mithun Prasad
Dr. Mitu Shrikhande
Dr. Mitul Shah
Dr. Mitusha Verma
Dr. Mohamed Ibrahim B K [MBBS, MS - General Surgery]
Dr. Mohammad Asim Siddiqui [MRCP (ENDOCRINOLOGY & DIABETES), MD (INTERNAL MEDICINE), MRCP(UK)(LONDON)]
Dr. Mohammed Abdel-Rahim Karajeh [Gastroenterology and Hepatology, Hepatobiliary ]
Dr. Mohammed Idhrees [MBBS, MS - General Surgery, MCh - Cardiothoracic Surgery, FAIS]
Dr. Mohammed Umar [MBBS, MD - Paediatrics]
Dr. Mohammed Umar [MBBS, MD - Paediatrics]
Dr. Mohan A Gadam
Dr. Mohan Chand Seal
Dr. Mohan Desai
Dr. Mohan Keshavamurthy
Dr. Mohan Kumar Singh
Dr. Mohd Irfan Banday [MBBS, MS - Orthopedics]
Dr. Mohd Irtaza [MBBS, MD Internal Medicine, DM Gastroenterology]
Dr. Mohd Qaleem
Dr. Mohit Bhardwaj [MBBS, MD - Pulmonology]
Dr. Mohit Khirbat [MBBS, MD - General Medicine, DNB - Nephrology]
Dr. Mohit Saxena [MBBS MD-General Medicine, DM-Medical Oncology]
Dr. Mohit Sharma [MBBS, MS , MCh , Hand Surgery Fellowship, Burns Fellowship]
Dr. Mohit Tyagi [MBBS, MD - Neonatology & Paediatrics]
Dr. Mohnish Tripathi [MBBS, DNB, IDCCM - Emergency Medicine]
Dr. Mohsin Khan
Dr. Monica Bambroo [MBBS, Diploma - Venerology and Dermatitis, Fellowship - Dermatology]
Dr. Monica Chib [MBBS, MD (Psychiatry)]
Dr. Monika Bhatia
Dr. Monika Meena
Dr. Monika Sharma [MBBS, MD - Medicine, DNB - Endocrinology]
Dr. Movva Srinivas
Dr. Mridul Chandra Das [DM, MD, DNB, MBBS - Gastroenterology]
Dr. Mridul Chandra Das [MBBS, MD, DM - Paediatric Gastroenterology]
Dr. Mridul Malhotra [MBBS, MD - General Medicine, DM - Medical Oncology]
Dr. Mridul Seth [Oral & Maxillofacial Surgery and Implantology]
Dr. Mridula Sarda
Dr. Mrigank Shekhar Jha [MBBS, MS - General Surgery, MCh - Urology]
Dr. Mriganka Juneja
Dr. Mriganka Sekhar Sharma [MBBS, MS - General Surgery, Fellowship - Bariatric and Upper GI Surgery]
Dr. Ms Upasana Mukherjee
Dr. Mudassar Kharadi [DNB (General Medicine), DNB (Neurology), Fellowship in Epilepsy]
Dr. Mukesh Goel [MBBS, MS - General Surgery, MCh - Cardio Thoracic Surgery]
Dr. Mukesh Kumar
Dr. Mukesh Kumar [MBBS ,MD Medicine, DM Neurology -AIIMS Delhi]
Dr. Mukesh Patekar[MBBS, MD - Internal Medicine, DM - Medical Oncology]
Dr. Mukti Mukherjee
Dr. Mukul Mutatkar [Diploma in Radiological Diagnosis]
Dr. Mukul Varma [MBBS, MD, DM]
Dr. Muneendra Gupta
Dr. Munia Bhattacharya [MA in Clinical Psychology, MPhil, RCI Certified Rehabilitation Psychologist]
Dr. Munish Paul
Dr. Murarji Ghadge [MBBS, DORL]
Dr. Murugan Natrajan
Dr. Muthu Veeramani [MBBS, MS - General Surgery, MCh - Urology]
Dr. Muzammil Shaikh
Dr. N S Saradha
DR. N. SUBRAMANIAN
Dr. Nabanita Bagchi Chatterjee [MBBS, MS (General Surgery), DNB (General Surgery), FNB (Minimal Access Surgery)]
Dr. Nabarun Roy
Dr. Nachiket Purandare [MS General Surgery, MCh Vascular Surgery]
Dr. Nagesh Chandra
Dr. Nagesh Sirsath [MD Medicine, DM Medical Oncology]
Dr. Naginder Vashisht [MBBS, MD - Ophthalmology, FRCS - Ophthalmology ]
Dr. Nagraj G. Huilgol
Dr. Nagraj S. Shetty
Dr. Nairyosan Irani [MS Ophthalmology]
Dr. Naman Utreja
Dr. Naman Wahal
Dr. Nameet Jerath [MBBS, MD Pediatrics, FIMSA, FIAP, FRCP (Glasgow), Fellow Pediatric Critical Care]
Dr. Namit Kalra Mohan
Dr. Namita Gupta
Dr. Namita Sharma
Dr. Nana Morkane
Dr. Nandini Hazarika
Dr. Narasimhan Subramanian
Dr. Narayan Jayashankar
Dr. Narendra S [MBBS, MS - General Surgery, MCh - Urology]
Dr. Naresh bansal
Dr. Naresh Kumar Goyal
Dr. NARESH MEHTA
Dr. Naresh Panwar [MBBS, MS, MCh - Neurosurgery]
Dr. NARESH SHETTY
Dr. Naresh Trehan
Dr. Narinder Tikoo [MBBS, MD]
Dr. Nasli Ichaporia [MBBS, MD - Neurology]
Dr. Naval Mendiratta [MBBS, Gold Medal in MD Medicine ]
Dr. Navdeep Singh Nanda
Dr. Naveen M A [MBBS, MS - General Surgery, MCh - Neuro Surgery, Neurointerventional Surgery]
Dr. Naveen Ravel [MBBS, MD - General Medicine, DM - Oncology]
Dr. Naveen Sanchety [MBBS, DNB - General Surgery, DNB - Surgical Oncology]
Dr. Naveen Saraf
Dr. Navin Chobdar
Dr. Navin Chobdar
Dr. Neel Kamal Sourav [MBBS, DNB - Orthopaedics]
Dr. Neel Shah [MBBS, DNB, FRCS]
Dr. Neelam Suri [MBBS,MD]
Dr. Neelima Limaye [MD Medicine]
Dr. Neelkanth Karandikar [MBBS, MD Medicine, DM Cardiology, FISE]
Dr. Neena Khanna
Dr. Neepa T. Dave
Dr. Neeraj Awasthy [MBBS, MD - Pediatrics, FNB - Pediatric Cardiology]
Dr. Neeraj Bhalla
Dr. Neeraj Chaudhary [MBBS, MS - General Surgery, DNB - Gastroenterology]
Dr. Neeraj Dhingra [MBBS, MD, DNB Radiotherapy]
Dr. Neeraj Godara[ Orthopaedic Hand, Foot & Microsurgeon ]
Dr. Neeraj Gupta
Dr. Neeraj Gupta [MBBS, DNB - Orthopedic Surgery, MNAMS - Orthopaedics]
Dr. Neeraj Gupta [MBBS, MD - Pulmonary Medicine, Fellowship in Critical Care]
Dr. Neeraj Narayan Mathur [MBBS,MS-ENT, DNB,FRSM,MAMS,FAMS]
Dr. Neeraj Singh [MDS, ORAL & MAXILOFACIAL SURGEON]
Dr. Neeraj Srivasthav
Dr. Neeraj Teotia [MBBS (JN Medical College, Belgaum), MD - Paediatrics, Fellowship in Paediatric Hematology & BMT]
Dr. Neeraj Verma [BDS, MDS, ADV.C. (ORTHODONTICS), DENTAL IMPLANTS]
Dr. Neerav Goyal
Dr. Neerja Goel
Dr. Neeta Kejriwal [MBBS, MD - Pediatrics]
Dr. Neeta Shah
Dr. Neetu Mittal [MBBS, MD - Microbiology]
Dr. Neha Berry
Dr. Neha Bhandari
Dr. Neha Bharti
Dr. Neha Dubey
Dr. Neha Gupta
Dr. Neha Kumar
Dr. Neha Mutha [MBBS, MS General Surgery, FISCP]
Dr. Neha Rastogi
Dr. Neha Rathi
Dr. Neha Sood [MBBS, DNB - ENT, Fellowship - Cochlear Implantation]
Dr. Nidhi Dhariwal
Dr. Nidhi Jain [MBBS, MS - Obstetrics & Gynaecology]
Dr. Nidhi Nayyar
Dr. Nidhi Rawal [MBBS, FNB - Pediatric Cardiology, MD - Pediatrics]
Dr. Nidhi Rohatgi [MBBS, Diploma in Dermatology]
Dr. Nidhi Sharma [MBBS, MD, MRCOG-1]
Dr. Nidhi Yadav [BDS, MDS]
Dr. Niharendu Ghara
Dr. Nikhil Agarwal
Dr. Nikhil Agrawal
Dr. Nikhil Jain [MBBS, MS - Orthopaedics, Fellowship - Spine Surgery]
Dr. Nikhil Likhate [MBBS, DNB (Orthopaedics), MNAMS]
Dr. Nikhil Mehta
Dr. Nikhil Modi [MBBS, MD, FCCP, EDARM, IDCCM]
Dr. Nikhil Narayan Bante
Dr. Nikhil Pal
Dr. Nikhil Parchure [MBBS, MD, Fellowship]
Dr. Nikhil Rishikeshi [MBBS, DOMS]
Dr. Nikhil S. Sardar
Dr. Nikhil Valsangkar [MBBS, DNB, MS - Orthopaedics]
Dr. Nikhil Yadav [MBBS, MS - General Surgery]
Dr. Nilesh Doctor [MBBS, MS - General Surgery, DNB - General Surgery]
Dr. Nilesh G Satbhai
Dr. Nilesh Jagtap [MBBS, MS - Orthopaedics]
Dr. Nipanjan Ghosh
Dr. Nipun Bajaj
Dr. Nipun Saha
Dr. Niraj Kumar [MBBS, MD, DNB - Interventional Radiology]
Dr. Niraj Vora
Dr. Niranjan Hiremath
Dr. Nirmal Raut
Dr. Nischol Raval [MBBS, DPM, FIPS]
Dr. Nisha Hariharan [MBBS, MS - General Surgery, Fellowship - Breast Oncology]
Dr. Nisha Rawat [BPT, MPT - Physiotherapy]
Dr. Nishant Rana [MBBS. MS ENT]
Dr. Nishant Soni [MBBS, MS - Orthopaedics, Fellowship - Hand & Microvascular Surgery]
Dr. Nishanth Sampath
Dr. Nishith Chandra
Dr. Nishtha Gupta [BDS, MDS - Orthodontics]
Dr. Nitasha Singh [MBBS, DNB - Medicine]
Dr. Niteen Kumar
Dr. Nitesh Rohatgi
Dr. Niti Kautish [MBBS, DNB - Obstetrics & Gynecology]
Dr. Nitika Kaur [BDS]
Dr. Nitika Nijhara
Dr. Nitika Sobti [MBBS, Diploma - Obstetrics and Gynaecology, Diploma - Minimal Access Surgery]
Dr. Nitin Annarapu
Dr. Nitin Arora [MBBS, MS - General Surgery, Fellowship in Robotic & Bariatric Surgery]
Dr. Nitin Goel[MBBS, MS - General Surgery, MCh - Paediatric Surgery]
Dr. Nitin Jagasia [MBBS, Fellowship - Emergency Medicine]
Dr. Nitin Kumar Parashar [MBBS, MD - Medicine, DM - Cardiology]
Dr. Nitin Pai [MD Medicine, DM Gastroenterology]
Dr. Nitin Pathak [MBBS, DNB (General Surgery), FCPS, DNB (Vascular & Endovascular Surgery)]
Dr. Nitin Pote [MBBS, MS - General Surgery]
Dr. Nitin Rathod
Dr. Nitin S. Walia
Dr. Nitin Shah [MBBS, MD - Paediatrics, DCH]
Dr. Nitin Shrivastava [MBBS, MS - General Surgery, MCh - Urology/Genito-Urinary Surgery]
Dr. Nitin Tiwari
Dr. Nitin Vashistha [MS, FIAGES, FACS (USA)]
Dr. Nitish Jhawar [MBBS, MS - General Surgery, Fellowship - Minimal Access Surgery]
Dr. Noaline Sinha [MBBS, Diploma in Radiation Medicine, Nuclear Medicine Physician]
Dr. Noor Ul Din Malik [MBBS MS (ENT)]
Dr. Nooreyezdan Shahin [MBBS, M.S, M.Ch(Plastic Surgery)]
Dr. Nupur Bit [MS General Surgery, MCh Vascular Surgery]
Dr. Nutan Agarwal [MBBS, MD - Obstetrics & Gynaecology]
Dr. NUTAN DESAI
Dr. O P Yadava [MBBS, MS, DNB(CTVS)]
Dr. Om Prakash Sharma [M.D.]
Dr. Oshin Bansal [DNB Ophthalmology, Diploma Ophthalmology]
Dr. P Balaji
Dr. P C Mondal
Dr. P K Das
Dr. P K Dave
Dr. P M Gopinath [MBBS, DGO, MD - Obstetrics & Gynaecology]
Dr. P N Mohapatra
Dr. P Nataraj [MBBS, MS - ENT]
Dr. P Nataraj [MBBS, MS - ENT]
Dr. P S Ashok Kumar [MBBS, MS - Orthopaedics Surgery, Orthopedic, Arthroplasty, Robotics & Navigation Joint Surgeon]
Dr. P Sridhar
Dr. P Suryanarayan
Dr. P Venkata Krishnan [MBBS, MD - Medicine]
Dr. P. Archana Rao
Dr. P. K. Purushothaman
Dr. P. Lakshmi Narasimman
Dr. P. M Purohit
Dr. P. Padmakumar
Dr. Padam Yadav [MBBS, MD - Pediatrics]
Dr. Padma Dudeja [MBBS, MD - Obstetrics and Gynecology]
Dr. Padmapriya Chandran
Dr. Pakhee Aggarwal
Dr. Pallav Kumar
Dr. Pallavi Sharma
Dr. Pallavi Vasal [MBBS, MD - Obstetrics & Gynaecology, Fellowship in Gynae Endoscopy]
Dr. Pankaj Aneja
Dr. Pankaj Bajaj [MBBS, MS - Orthopaedics, DNB - Orthopedics/Orthopedic Surgery]
Dr. Pankaj Gunjal [MBBS, DNB (Orthopaedics), Diploma in Orthopaedics]
Dr. Pankaj Khatana [MBBS, MD - Internal Medicine, Diabetology Training]
Dr. Pankaj Kumar Gupta [MBBS, MD, FNB, FIACTA - Anaesthesia]
Dr. Pankaj Lohia
Dr. Pankaj Magar [MBBS, DTCD, DNB (Chest & Pulmonology)]
Dr. Pankaj Maheshwari
Dr. Pankaj Mehta
Dr. Pankaj Panwar
Dr. Pankaj Puri
Dr. Pankaj R. Shroff
Dr. Pankaj Sugaonkar [DCH, DNB (Paediatrics), FNB (Paediatric Cardiology)]
Dr. Pankaj Sugaonkar [MD Paediatrics, DM Paediatric Cardiology]
Dr. Pankaj Walecha [MBBS, MS - Orthopaedics, Robotic Surgery]
Dr. Parag Kumar
Dr. Parag Sahasrabudhe [MBBS, MS, MCh - Plastic Surgery]
Dr. Parag Vibhakar
Dr. Paras Agarwal [MBBS, MD, FRCP (Glasgow), FRCP (Edinburgh)]
Dr. Paras Satadeve [MBBS, MD, DM - Clinical Hematology, EHA Certification]
Dr. Pardeep Bageja
Dr. Pardeep K Bansal
Dr. Paresh Jain
Dr. Parimal Kore [D Ortho, DNB Ortho]
Dr. Paritosh Pandey
Dr. Paritosh S. Gupta [MBBS, MS - General Surgery, DNB - General Surgery, Robotic Surgery]
Dr. Parjeet Kaur [MBBS, MD - Medicine, DM - Endocrinology]
Dr. Parth Vadhadia [MBBS, MD, DrNB - Gastroenterology]
Dr. Parthasarathy G
Dr. Parul Garde
Dr. Parul Katiyar
Dr. Parul Prakash [MBBS, MD - Obstetrics & Gynaecology, FNB - Reproductive Medicine]
Dr. Parul Sharma
Dr. PARUL TANK
Dr. Parvathi Unninayar
Dr. Parveen Jain [MBBS, DNB - General Medicine, DNB - Medical Oncology]
Dr. Parveen Yadav
Dr. Patta Radhakrishna
Dr. Pawan Goyal[MBBS, MS - General Surgery, MCh - Neuro Surgery]
Dr. Pawan Gupta
Dr. Pawan Kumar Singh[Haemato-oncologist & Bone Marrow Transplant]
Dr. Pawan Rawal [MBBS, MD - Pediatrics, DM - Gastroenterology]
Dr. Pawanindra Lal [MBBS, MAMC, MS Surgery,DNB Surgery FRCS (Edinburgh, FRCS Glasgow , FRCS England , FRCS Ireland ), FACS, FAMS, FFST Edinburgh]
Dr. Peeyush Jain
Dr. Peeyush Kumar
Dr. Peeyush Tomar [MBBS, DrNB - Neurosurgery, Fellowship - Stroke and Neuroendovascular Intervention]
Dr. Peush Bajpai [MBBS, MD - Medicine, DM - Medical Oncology]
Dr. Phulchand Pujari [MBBS, DNB - General Surgery, Fellowship - Minimal Access Surgery]
Dr. Piyush Gupta [ MBBS, MS - General Surgery, MCh - Urology, Robotic Surgery Specialist]
Dr. Piyush Kumar Agrawal [MBBS, MS - General Surgery, MCh - Surgical Oncology]
Dr. Piyush Lodha [MD Medicine, DM Endocrinology]
Dr. Piyush Marudwar
Dr. Piyush Ojha [MBBS, MD - Internal Medicine, DM - Neurology, Fellowship in Interventional Neurology (Canada)]
Dr. Piyush Singhania [MBBS, MS - General Surgery, MCh - Urology]
Dr. Piyush Vishwakarma [MBBS, MD, DrNB]
Dr. Pooja Aggarwal [MD - Dermatology and Venerology, MBBS]
Dr. Pooja Banerjee
Dr. Pooja Wadwa [MBBS, MD - Anaesthesiology, IDCCM]
Dr. Poornima Sivakumar
Dr. Prabash Prabhakaran
Dr. Prabhat Maheshwari [MBBS, MD - Pediatrics, DNB - Pediatrics, Paediatrician and Neonatology]
Dr. Pradeep Alate [MRCP, DCH]
Dr. Pradeep B. Bhosale
Dr. Pradeep Champawat [MBBS, MS, DNB Urology]
Dr. pradeep chowbey
Dr. Pradeep Divate [MBBS, MD (Internal Medicine), DM (Neurology) - NIMHANS]
Dr. Pradeep Jain [MBBS, MD, DM]
Dr. Pradeep Kumar Moolchandani [MBBS, MD, DM - Critical Care]
Dr. Pradeep Kumar Singh [MBBS, MS - General Surgery , MCh - Burn & Plastic Surgery]
Dr. Pradeep Moonot [MBBS, DNB Orth, MS Orth, MRCS, FRCS]
Dr. Pradeep Muley
Dr. PRADIP SHAH
Dr. Pradyumna J. Oak
Dr. Praful Kamani [MBBS, MD - Internal Medicine, DNB - Gastroenterology]
Dr. Prafulla Tamaskar [MS (General Surgery)]
Dr. Pragya Khare
Dr. Pragya Rai
Dr. Prajakta Mehendale
Dr. Prakash M. Doshi
Dr. PRAKASH VAIDYA
Dr. Pramod Kadam [MS General Surgery]
Dr. Pramod Krishnan [MBBS, MD - General Medicine, DM - Neurology]
Dr. Pramod Kumar Jaiswal
Dr. Pramod Kumar Julka
Dr. Pramod Narkhede [DNB Medicine, DNB Cardiology]
Dr. Pramoj Jindal [MBBS, DNB - Laparoscopic Surgery, Clinical Fellowship - Thoracic Surgery and Thoracic Onco-surgery]
Dr. Pranav Jadhav [MBBS, MS, M.Ch (Paediatric Surgery)]
Dr. Pranav Kumar [MBBS, MS, MCh]
Dr. Pranav Rajesh Agrawal
Dr. Pranav Thusay [DNB General Surgery, MCh Plastic & Reconstructive Surgery]
Dr. Pranjal Pandit [MBBS, DNB General Surgery, MCh Plastic Surgery]
Dr. Prasad Bhagunde
Dr. Prasad Suryawanshi [MD Anaesthesiology, DNB Anaesthesiology, FNB Critical Care]
Dr. Prasan Deep Rath [MBBS, MD - Internal Medicine, Fellowship - Rheumatology]
Dr. Prasanna Kumar K S
Dr. Prasenjit Chatterjee
Dr. Prashant Agrawal [MBBS, MS - Orthopaedics, DNB - Orthopedics]
Dr. Prashant Baid
Dr. Prashant Bhardwaj [MD - Physician, FCCM]
Dr. Prashant Chhajed
Dr. Prashant Kumar
Dr. Prashant Mehta
Dr. Prashant Pawar
Dr. Prashant Pawar [MBBS, MS - ENT, Diploma - Otorhinolaryngology]
Dr. Prashant Saxena
Dr. Prashant Udavant [MD Paediatrics]
Dr. Prashant Vanzar [MBBS, DNB General Surgery, DNB Surgical]
Dr. Prashant Vasant Bhamare
Dr. Prashanta Kumar Ghosh [MBBS, MD(Med), DM(Cardiology)]
Dr. Prasun Chatterjee
Dr. Pratap Varma Penmetsa
Dr. Prateek Vardhan [MBBS, MS, FIAGES, FALS - General Surgery]
Dr. Pratibha Gogia
Dr. Pratibha Singhi
Dr. Pravas Chandra Mishra [MBBS, MD-Internal Medicine, DM-Clinical Hematology]
Dr. Praveen Chandra
Dr. Praveen Gupta [MBBS (University of Nagpur, 1998), MD - General Medicine (Maulana Azad Medical College, Delhi, 2002 - Distinction), DM - Neurology (AIIMS New Delhi, 2005 - Gold Medalist)]
Dr. Praveen Kulkarni
Dr. Praveen Kumar Bansal MBBS, MD (Medicine) Gold Medalist, DM (Medical Oncology)
Dr. Praveen Patil [MRCP, Fellowship in Rheumatology]
Dr. Praveen Sodhi [MBBS, MS Laproscopic, Gastroenterology, Anorectal & Breast Surgery]
Dr. Praveer Agarwal
Dr. Pravin Ganjre [MBBS, MS, MCh Neurosurgery, MCNS USA]
Dr. Pravin Jain [MBBS, DNB (Neurosurgery), DNB (General Surgery)]
Dr. Preetam Krishnamurthy
Dr. Preetha Joshi [MBBS, DCh, FCPS, DNB Pediatrics]
Dr. Preethi Jeyaraman
Dr. Preethiswary Subramanian
Dr. Preeti Pandya [MBBS, MS - General Surgery, MCh - Plastic Surgery (Gold Medal)]
Dr. Preeti Singh [BDS, MDS, FAGE]
Dr. Preeti Vijayakumaran [ MBBS – Gold Medalist, MS - General Surgery, MCh - Surgical Oncology]
Dr. Preetinder Singh Lamba
Dr. Premal Narkhede
Dr. Premanand N
Dr. Premchand
Dr. Prita Trehan [MBBS, MD(Paediatrics)]
Dr. Pritha Rakshit
Dr. Pritish Singh [MBBS, MS - Orthopaedics]
Dr. Priya Jain [MD(Pediatrics), DNB(Pediatrics), PG DDN(Developmental Neurology)]
Dr. Priya Sharma [MBBS, DNB, DM - Pulmonary Medicine]
Dr. Priya Sharma [MBBS, DNB, DM - Pulmonology]
Dr. Priya Tiwari [MBBS Gold Medalist, MD - General Medicine, DM - Medical Oncology]
Dr. Priyadarshi Amit
Dr. Priyanka Hemrajani [MBBS, MD - Dermatology, International Training in Cosmetic Dermatology]
Dr. Priyanka Pandurang Navelkar
Dr. Priyanka Pipara
Dr. Priyanka Raina [MDS (Oral & Maxillofacial Surgery RGUHS)
Dr. Priyanka Sharma [MBBS, MD - Microbiology]
Dr. Priyanka Singh [MBBS, MD - Internal Medicine]
Dr. Priyanka Sinha
Dr. Priyanka Trivedi
Dr. Prof Col Pradyot Sarkar
Dr. Prof Dhavendra Kumar [MBBS, MD, DCH (RCPSI), MMedSci, DABMG, FRCPI, FRCP, FRCPCH, FACMG, DSc. (Hon)]
Dr. Prof Koushik Lahiri
Dr. Prof Narendra Nath Khanna [MD, DM, MNAMS, FACC (USA), FSCAI (USA), FCCP (USA), FESC, FAPSIC, FIMSA. FEISI, FICC]
Dr. Prof Rajesh Malhotra [M.B.B.S (AIIMS, New Delhi), M.S Orthopedics (AIIMS, New Delhi), F.R.C.S (Glasgow) FACS, FICS, FIMSA, FNAMS, FNASc.]
Dr. Prof Raju Vaishya [MBBS, MS (Orth), MCh (Liverpool), FRCS (London), FACS (USA), PG DHA]
Dr. Prof Rohini Handa [MBBS, MD, DNB, FAMS, FICP, FACR, FRCP (Glasgow)]
Dr. Prof Suresh Singh Naruka [MBBS (Medicine),MS (Otorhinolaryngology, and Head & Neck Surgery)]
Dr. Prof. Uday C Ghoshal
Dr. Puja Kapoor [MBBS, DNB - Paediatrics, Fellowship - Paediatric Neurology]
Dr. Puja Rai [MBBS, MS, DNB, FAICO - Ophthalmology]
Dr. Pulak Vatsya
Dr. Puneet Agarwal
Dr. Puneet Ahluwalia
Dr. Puneet Bedi [MBBS, MD]
Dr. Puneet Dwevedi [M.D. (Psychiatry), MBBS, Mental Health and Behavioural Sciences]
Dr. Puneet Girdhar
Dr. Puneet Khanna [MBBS, MD - Respiratory Medicine, Fellowship]
Dr. Puneet Varma
Dr. Punit L Jain [MBBS, MD - General Medicine, DM - Clinical Haematology]
Dr. Punit Singla
Dr. Purneshwar Kumar Pandey [MBBS, MD - General Medicine, DM - Cardiology]
Dr. Purshotam Lal
Dr. Purushottam Vashistha [MBBS, MD - General Medicine, DM - Gastroenterology]
Dr. Purvez K. Grant [MBBS, MD, FACC, FSCAI, FRSM, FAPSIC, FCSI]
Dr. Purvi Kadakia Kutty [MBBS, MD - Paediatrics, FNB - Pediatric Hematology and Oncology]
Dr. Pushkar Ramesh Ingle
Dr. Pushpendra Nath Renjen [MBBS, DM (Neuro) FRCP (Glas.), FRFCP (Edin), FRCP (Ireland).]
Dr. Pushpinder Gulia
Dr. R Krishnamoorthy [MBBS, MS - General Surgery, MCh - Plastic Surgery]
Dr. R N Srivastava [MBBS, Diplomate of the American Board of Paediatrics]
Dr. R Ravichandran [MBBS, MD - General Medicine]
Dr. R Ravichandran [MBBS, MD - General Medicine]
Dr. R Sundararaman [MBBS, MD - General Medicine]
Dr. R Sundararaman [MBBS, MD - General Medicine]
Dr. R Sunil Kapilavayi
Dr. R. Balaji
Dr. R. K. Chopra [MD Medicine, MD Respiratory Medicine, DTCD, FCCP]
Dr. R. Nagarajan [MS, MCh]
Dr. R.C.M Kaza
Dr. R.k Pandey
Dr. R.K Thapar [MBBS MD - Pediatrics]
Dr. Raashidha [MBBS, MD - Obstetrics & Gynaecology]
Dr. Raashidha [MBBS, MD - Obstetrics & Gynaecology]
Dr. RACHANA DEEPAK TATARIA
Dr. Rachit Agrawal [MBBS, MD - Internal Medicine, DM - Clinical Hematology]
Dr. Rachna Jagia
Dr. Radhika Chavan [MBBS, MD (Internal Medicine), DrNB (Medical Gastroenterology)]
Dr. Radhika Ruhatiya
Dr. Rafat Trivedi [MBBS, MD - Paediatrics, Fellowship in Paediatric Neurology]
Dr. Raghav Barve [MS Ortho, DNB Ortho, Fellow Oxford UK]
Dr. Raghav Kapoor
Dr. Raghavan L
Dr. Raghavan Samudrala
Dr. Raghavi Vishnu Prasanna
Dr. Raghunandan Prasad [MBBS,MD-Radio-diagnosis]
Dr. Rahil Chaudhary [MBBS, MS - Ophthalmology(Gold Medalist)]
Dr. Rahul Bhargava [MBBS, MD - General Medicine, DM - Clinical Haematology]
Dr. Rahul Chandhok [MBBS, MD- Psychiatry, Mental Health and Behavioural Specialist]
Dr. Rahul Chowdhary
Dr. Rahul Deshpande [MBBS, DOMS, MS (Ophthalmology), DNB (Ophthalmology)]
Dr. Rahul Gupta
Dr. Rahul Gupta [MBBS, MS - General Surgery, MCh - Neuro Surgery]
Dr. Rahul Gupta [MBBS, MS - Orthopaedics]
Dr. Rahul Kapoor [M.Ch-Plastic Surgery, MS-Gen Surgery, MBBS]
Dr. Rahul Magar [MBBS, DORL (ENT), MBA (Hospital Administration)]
Dr. Rahul Manchanda [MBBS, MD - Obstetrics & Gynaecology]
Dr. Rahul Mehrotra [MBBS, MD - Medicine, DNB - Cardiology]
Dr. Rahul Misra [MBBS, MD, FRCR Clinical Oncology Fellowship]
Dr. Rahul Nagpal
Dr. Rahul R Gupta [MBBS, MD - General Medicine, DM - Cardiology]
Dr. Rahul Sawant [MD, MRCP, CCT]
Dr. Raina Chawla
Dr. Raina N. Nahar
Dr. Raj Kumar
Dr. Raj Kumar Jain
Dr. Raj Kumar [MBBS, MD - General Medicine]
Dr. Raj Kumar [MBBS, MD - General Medicine]
Dr. Raja Vikram Saikia
Dr. Rajan Madan
Dr. Rajan Shah
Dr. Rajat Agrawal
Dr. Rajat Ahluwalia
Dr. Rajat Bhattacharyya
Dr. Rajat Goel
Dr. Rajeev Bhardwaj [MBBS, MD - Anaesthesiology]
Dr. Rajeev Chaudhari [MS General Surgery, MCh Urology]
Dr. Rajeev Kumar Rajput [MBBS, MD - General Medicine, DM - Cardiology]
Dr. Rajeev Kumar [MBBS, MD - Radiology]
Dr. Rajeev Shandil [MBBS, DNB(med), DNB(Gastro) Fellowship EUS, MANOMETRY, ENDOBARIATRICS]
Dr. Rajeev Sivasankar [Neurovascular & Peripheral interventional radiology]
Dr. Rajeev Thilak Chellasamy
Dr. Rajeev Verma
Dr. RAJEN DOSHI
Dr. Rajendra Fiske [MBBS , DNB Orthopaedics, Diploma Orthopaedics]
Dr. Rajendra M. Saraogi
Dr. Rajendra Patil [MBBS, MD - General Medicine, DNB - Cardiology]
Dr. Rajendra Prasad
Dr. Rajesh Ahlawat [MBBS, MS - General Surgery, MCh - Urology]
Dr. Rajesh Benny
Dr. Rajesh Ganatra
Dr. Rajesh Goel
Dr. Rajesh Jha [MBBS, DNB - Paediatrics]
Dr. Rajesh K Chawla
Dr. Rajesh Kaushish [MBBS, MS (General Surgery), M.Ch (CVTS)]
Dr. Rajesh Kaushish [MBBS, MS - General Surgery, MCh - Cardio Thoracic Surgery]
Dr. Rajesh Kumar Jain [MBBS, MS - General Surgery, DNB - Surgical Oncology]
Dr. Rajesh Kumar Pande
Dr. Rajesh Kumar Verma [MS - Orthopaedics, MBBS]
Dr. Rajesh Kumar Watts
Dr. Rajesh Nandan Sahay [MBBS, Dip. Cardiology, FICCM]
Dr. RAJESH NATHANI
Dr. Rajesh S. Parasnis [MBBS, MS Ortho, DNB Ortho, Fellowsip in Spine Surgery,]
Dr. Rajesh Shinde [MBBS, MS- General Surgery, MCh - Surgical Oncology]
Dr. Rajesh Taneja
Dr. Rajesh Thachathodiyl [MBBS, MD - General Medicine, DM - Cardiology]
Dr. Rajesh Valeja [MBBS, MS - General Surgery]
Dr. Rajesh Verma
Dr. Rajeshwar Balkrishna [MBBS, DGO, MD Ob-Gyn]
Dr. Rajeswari Sendhil
Dr. Rajeswari Thangavel
Dr. Rajiv Agarwal
Dr. Rajiv Anand
Dr. Rajiv Chhabra[MBBS, MD - Pediatrics, Paediatrics and Neonatology]
Dr. Rajiv Dang
Dr. Rajiv G. Bhagwat
Dr. Rajiv Karnik
Dr. Rajiv Lochan J
Dr. Rajiv Mohan
Dr. Rajiv Yadav [MBBS, MS - General Surgery, MCh - Urology]
Dr. Rajkamal Vishnu [MBBS, MS - General Surgery, MCh - CTVS]
Dr. Rajkumar H. Shah
Dr. Rajneesh Malhotra
Dr. Rajni Farmania
Dr. Rajnish Kumar [DM - Neurology, MD - General Medicine, MBBS]
Dr. Rajnish Sardana
Dr. Raju P Kubchandani
Dr. Rajul Aggarwal
Dr. Rakesh Agarwal
Dr. Rakesh Gupta [MBBS, MD(Medicine), FIACM]
Dr. Rakesh Gupta [MD - Internal Medicine, MBBS]
Dr. Rakesh Jalali
Dr. Rakesh Jha [MBBS]
Dr. Rakesh Kumar Khazanchi [MBBS, MS - General Surgery, MCh - Plastic Surgery]
Dr. Rakesh Mahajan
Dr. Rakesh Neve [MBBS, MS - General Surgery, Diploma - Gynaecologic Laparoscopy]
Dr. Rakesh Rai Sapra
Dr. Rakhi Anand [Ph.D Clinical Psychology]
Dr. Rama Joshi [Gold Medalist MBBS , MS - Obstetrics & Gynaecology, Robotic Surgery]
Dr. Ramakanta Panda
Dr. Ramakanta Panda [MBBS, MCh - Cardio Thoracic and Vascular Surgery]
Dr. Raman Gaikwad [MBBS, MD - Internal Medicine, FNB - Infectious Diseases]
Dr. Raman Gaikwad [MD, FNB (Infectious Diseases)]
Dr. Raman Kant Aggarwal
Dr. Raman Kumar Sharma
Dr. RAMAN MALIK
Dr. Raman Puri [MD, DM]
Dr. RAMAN SHENOY
Dr. Raman Tanwar [MBBS, MCh - Urology, MS - General Surgery]
Dr. Ramandeep Singh Arora
Dr. Ramani Narasimhan [Fellow - Pediatric Orthopaedic Surgery, MS(Orthopaedic Surgery), MBBS]
Dr. Ramdip Ray
Dr. Ramdip Ray
Dr. Ramesh C. Sharma
Dr. Ramesh L. Rohiwal [MS ENT]
Dr. RAMESH MAHAJAN
Dr. RAMESH PUNJANI
Dr. Ramesh Sarin
Dr. Ramji Mehrotra [MBBS, MS - General Surgery, MCh - Cardio Thoracic Surgery]
Dr. Ramkinkar Jha [MBBS, MS - Orthopaedics, Orthopaedics & Joint Replacement Surgeon]
Dr. Ramkishan Nag
Dr. RAMKRISHNAN MODI
Dr. Ramna Banerjee
Dr. Ramneek Mahajan [MBBS, MS - Orthopaedics]
Dr. Rana Patir
Dr. Randeep Guleria [DM - Pulmonary Medicine, MD - Medicine, MBBS]
Dr. Randhir Kenjale [MBBS, D Ortho, FCPS Ortho]
Dr. Randhir Sud
Dr. Ranjan Kamilya
Dr. Ranjan Kumar [MBBS, MD - Pathology]
Dr. Ranjan Kumar [MBBS, MD - Pathology]
Dr. Ranjan Shetty
Dr. Ranjana Mithal [MBBS, MS]
Dr. Ranjana Sharma [MBBS, MS, MRCOG(UK), FROG (UK), MFFP (UK), FICOG, PG Cert in Med Edu]
Dr. Ranjeet Deshmukh [MBBS, DNB (Neurosurgery)]
Dr. Ranjeet Deshmukh [MBBS, MS - General Surgery, MCh - Neurosurgery]
Dr. Ranjit Ghuliani [MBBS, MD Pediatrics]
Dr. Rashida Patanwala Melinkeri [DNB (Internal Medicine)]
Dr. Rashmi Aderao [MBBS, MD Skin, VDMD]
Dr. Rashmi Algeri
Dr. Rashmi Bhamare [MBBS, MS - Obstetrics & Gynaecology, DNB - Obstetrics & Gynecology]
Dr. Rashmi Bhardwaj [MBBS, MD - Paediatrics, Fellowship in Neonatology & PICU]
Dr. Rashmi Parikh
Dr. Rashmi Sharma [DNB - Dermatology & Venereology, MBBS, Diploma in Dermatology, Venereology and Leprosy]
Dr. Rashmi Taneja [MBBS, Diplomate of the American Board of Plastic Surgery]
Dr. Rashmie Prabha [MBBS, MD - Paediatrics]
Dr. Ratanadip Bose [MBBS, MS - General Surgery, MCh - Neurosurgery]
Dr. Rathan Shetty
Dr. Ratnav Ratan
Dr. Ratnesh Kumar Jaiswal [MBBS, DNB, FMAS, FALS, FIAGES]
Dr. Ravi Bhatia
Dr. Ravi Bhatia [MBBS, Diploma in Otorhinolaryngology (DLO)]
Dr. Ravi Ganesh Bharadwaj
Dr. Ravi Gopal Varma
Dr. Ravi Kale [MBBS, MCh - Paediatric Surgery, MS - General Surgery]
Dr. Ravi Kiran Barigala
Dr. Ravi Kumar Joshi
Dr. Ravi Kumar Shah [MBBS, MD - Internal Medicine, Fellowship in Geriatric Medicine]
Dr. Ravi Sauhta [MBBS, MS - Orthopaedics, MCh - Orthopedics, Orthopaedics & Joint Replacement Surgeon ]
Dr. Ravi Shekhar Jha
Dr. Ravikant Arora [MBBS, MS - General Surgery]
Dr. Ravikiran Muthuswamy
Dr. Ravindra Ghule
Dr. Ravindra Gulati [MBBS, MD, FACC, FACP]
Dr. Ravindra Kulkarni [MBBS, MD (Medicine), DNB (General Medicine), FSCAI]
Dr. Ravindra Kulkarni [MBBS, MD - Internal Medicine, DNB - Cardiology]
Dr. Ravindra Nikalji [MBBS, MD - Internal Medicine, DM - Nephrology]
Dr. RAVINDRA RUPWATE
Dr. Ravindra Saxena
Dr. Ravindra Singh
Dr. Ravindra Srivastava [Gold Medalist in MBBS, MCh - Neuro Surgery MS - General Surgery]
Dr. Ravindra Vats
Dr. Ravindran R
Dr. Raviraj Sinh Gohil [MBBS, MDS, FCMFT, AOMSI Fellowship]
Dr. Ravishankar R [MBBS, MS - ENT]
Dr. Ravishankar R [MBBS, MS - ENT]
Dr. Rayaz Ahmed [MBBS, MD - Pathology, DM - Clinical Haematology, Gold Medal in MD]
Dr. Reena Mittal [MBBS, MD - Pathology, Fellowship - Haematopathology]
Dr. Reena Shrotri [MBBS, DMRD]
Dr. Reetadyuti Mukhopadhyay
Dr. Reetesh Gupta [MBBS, MD]
Dr. Reetesh Ranjan
Dr. Rekha Ambegaokar
Dr. Rekha Jaiswal [MBBS, MS(General Surgery), FIAGES, FMAS, FALS(Bariatric), DMAS and Robotic (de Vinci)]
Dr. Renaldo Pavrey
Dr. Renu Gupta
Dr. Renu Raina Sehgal [MBBS, DNB - Obstetrics & Gynecology]
Dr. Reshma Puranik [MBBS, MD, DNB Medicine, DM, DNB Medical Oncology, MRCP UK]
Dr. Richa Bansal [MBBS, MS - Obstetrics and Gynaecology, MCh - Gynecological Oncology]
Dr. Richa Chaturvedi [Endocrinologist]
Dr. Richa Ranjan [Vitreo Retina Surgery]
Dr. Richa Sareen
Dr. Richa Singh [MBBS, Diploma - Obstetrics & Gynaecology, DNB]
Dr. Riddhijyoti Talukdar
Dr. Rina K
Dr. Rinkesh Kumar Bansal [MBBS, MD - Medicine, DNB - Gastroenterology and Hepatology]
Dr. Rishabh Jain
Dr. Rishabh Kedia
Dr. Rishabh Kumar
Dr. Ritesh Mongha [MBBS , DNB - General Surgery, MS - Surgery, MCh - Urology
Dr. Ritesh Narula
Dr. Rithesh R Nair [MBBS, MD - General Medicine, DM - Neurology]
Dr. Ritika Malhotra [BDS, MDS - Oral & Maxillofacial Surgery]
Dr. Ritika Sachdev
Dr. Ritu Jha
Dr. Ritu Jha
Dr. Rituparna Shinde [MD, DNB (Cardiology)]
Dr. Rituraj Baruah
Dr. Ritwick Raj Bhuyan
Dr. Rizwan Shaikh
Dr. Robert Coelho [MBBS, MS - General Surgery, MCh - Cardiothoracic and Vascular Surgery, Pediatric Cardiac Surgery]
Dr. Rochan Pant [MD Radio Diagnosis]
Dr. Rohan Aurangabadwalla [MBBS, MD - Respiratory Medicine]
Dr. Rohit Bansil
Dr. Rohit Chauhan [MBBS, MS, MCh - Urology]
Dr. Rohit Chauhan [MBBS, MS, MCh - Urology]
Dr. Rohit Goyal
Dr. Rohit Hegde
Dr. Rohit Kumar Garg
Dr. Rohit Lamba [MBBS, MS - Orthopedics, Fellowship - Hip and Knee Replacement]
Dr. Rohit Malhotra [MBBS, MD, DM - Cardiac Anaesthesia]
Dr. Rohit Nayyar
Dr. Rohit Om Parkash
Dr. Rohit Vohra [MBBS, MD PEDIATRICS, FELLOWSHIP IN PEDIATRIC INTENSIVE CARE,FELLOWSHIP IN PULMONOLOGY]
Dr. Roji Philip
Dr. Ronak Kaushik Sheth
Dr. Roohi Muhammad Yunus Khan
Dr. Roopa Salwan
Dr. Roopali Suryawanshi [MD Microbiology]
Dr. Ruchi Bhandari
Dr. Ruchi Goel [MBBS, MS, DNB, FAICO(Oculoplasty), FRCOphth(London)]
Dr. Ruchi Shrivastava [MBBS, MD - Obstetrics and Gynaecology, Fellowship - Gynaecologic Oncology]
Dr. Ruchi Shrivastava [MBBS, MD - Obstetrics and Gynaecology, Fellowship - Gynaecologic Oncology]
Dr. Rujuta Mehta
Dr. Rupal Chheda
Dr. Rupal Gupta [MBBS, MS - Ophthalmology]
Dr. Rupali Goyal [MBBS, DNB ( OBS & GYNAEC), Dip in Usg]
Dr. Rupali Sachdeva [MBBS, MD, DNB - Medicine]
Dr. Rupashree Dasgupta
Dr. Rupinder Sekhon [MBBS, MD, Diploma - Obstetrics and Gynecology, Robotic Gynaecological Oncology surgeries.]
Dr. Ruquaya Ahmad Mir [MBBS, DNB]
Dr. RVS Bhalla
Dr. S Chatterjee [MBBS, MD(medicine)]
Dr. S Hukku
Dr. S Jayaraman
Dr. S K Agarwal [MD, MNAMS, FICP]
Dr. S K Gupta [MD, DM, DIP.NB Cardiology, MNAMS, FICA (USA)]
Dr. S K Pal [MBBS. MS, M.Ch]
Dr. S N Singh
Dr. S Thiyagarajan
Dr. S V Lakshmi
Dr. S Velmurugan [MBBS, MS - Orthopaedics]
Dr. S Vidyadhara
Dr. S. Jayalakshmi [MBBS, MD - Radiotherapy, DNB - Radiotherapy, Specialty-Radiation Oncology & CyberKnife]
Dr. S. K. (Sushil Kumar) Kabra [MD (Ped), DNB (Pediatrics)Diplomat of Pediatric Respiratory Medicine (ERS)]
Dr. S. RadhaKrishnan
Dr. S. Sakthi Raja
Dr. S. Sathiyan
Dr. S.Bharti
Dr. S.V. Kotwal [MBBS, MS - General Surgery, MCh - Urology]
Dr. SABHSINGH KHAMBAY
Dr. Sabir Husain Ansari [MBBS, MS]
Dr. Sabrina Bokil [MD, DGO, FRCOG, MRCOG]
Dr. Sabyasachi Bandyopadhyay
Dr. Sachin Athalye [MS Ophthalmology]
Dr. Sachin Kandhari
Dr. Sachin Karkamkar [MBBS, MS (Orthopaedics)]
Dr. Sachin Karkamkar [MBBS, MS - Orthopaedics, Fellowship - Joint Replacement Surgery]
Dr. Sachin Kumar Jain
Dr. Sachin Mahajan [MBBS, MS (General Surgery), M.Ch (Neurosurgery)]
Dr. Sachin Maheshwari [MBBS, DA, DNB - Anaesthesia]
Dr. Sachin Mittal MBBS, MS - General Surgery, DNB - General Surgery, FNB - Minimal Access Surgery
Dr. Sachin Pathak [MBBS, D.Orth, Fellowship in Spine Surgery]
Dr. Sachin Patil [MBBS, MD - General Medicine, DM - Nephrology]
Dr. Sachin Tapasvi [MS (Orthopaedics), DNB (Orthopaedics), FRCS (Glasgow), MNAMS, AFAOA (Australia)]
Dr. Sachin Trivedi
Dr. Sachin Vaze [MBBS, DNB (General Surgery), DNB (Surgical Gastroenterology)]
Dr. Sadanand Dey
Dr. Sadanand Karandikar [MD, Fellowship Medical Oncology]
Dr. Sagar Walankar [MBBS, DNB - Paediatrics, Diploma - Allergy Asthma Immunology]
Dr. Sahil Arora [MBBS, MD Medicine, DM Nephrology, DrNB Nephrology, MNAMS, ESENeph (MRCPUK), FGN (WKA, USA), Fellowship in Intervention Nephrology(INU)]
Dr. Sahil bagai [DM - Nephrology, MBBS, MD - General Medicine]
Dr. Sahil Bansal [MBBS, MD - Radiodiagnosis, Fellowship - Vascular and Interventional Radiology]
Dr. Sahil Gaba
Dr. Sahil Rasane [DNB Medicine, DNB Gastroenterology]
Dr. Sahil Sohi [MBBS, DNB - Orthopedics/Orthopedic Surgery]
Dr. Sai Krishna B Naidu
Dr. Saibal Moitra
Dr. Saifuddin Bandukwala [MBBS, Fellowship, MD - General Medicine]
Dr. Sairam Subramanian
Dr. Sajal Ajmani [MBBS, MD - Medicine, MRCP (UK), DM - Clinical Immunology and Rheumatology]
Dr. Sajal Kamat [MBBS, MD (Internal Medicine), DM (Endocrinology)]
Dr. Sajjan Rajpurohit
Dr. Saket Bhardwaj [MBBS, MD - Medicine, DM - Cardiology]
Dr. Saket Goel [MBBS, MS (Surgery)]
Dr. Saket Sathe
Dr. Saksham Mittal
Dr. Sakshe Jain [MBBS, MD Psychiatry]
Dr. Sakshi Karkra [MBBS, MD - Pediatrics, Fellowship - Pediatrics, Gastroenterology and Hepatology]
Dr. Sakshi Mittal
Dr. Sakthi Saranya P. S.
Dr. Salil Patkar [MBBS, MD - General Medicine, DM - Oncology]
Dr. Salil S. Bendre
Dr. Salil Shirodkar
Dr. Samanjoy Mukherjee [MBBS, MD - General Medicine, DM - Cardiology]
Dr. Samarth Arya [MBBS, MS - Orthopaedics, DNB - Orthopaedics]
Dr. Sameer Ashok Rege
Dr. Sameer Bhargava [MBBS, MD – Psychiatry]
Dr. Sameer Bhate [MBBS, DNB, M.Ch, FIACS, Fellowship]
Dr. Sameer Desai [MBBS, MS (Orthopaedics), DNB, MRCS, D.Orth, Fellowship in Paediatric Orthosurgery]
Dr. Sameer Desai [MS Ortho, Fellowship Paediatric Ortho]
Dr. Sameer Kaul [MBBS, MS - Surgical Oncology]
Dr. Sameer Kaushal [MD - Ophthalmology(AIMS), DNB - Ophthalmology, MBBS]
Dr. Sameer Mehrotra [MBBS, DM - Cardiology, MD - General Medicine]
Dr. Sameer Pagad
Dr. Sameer Sadawarte
Dr. Sameer Shrivastava
Dr. Sameer Tulpule
Dr. Samir Parik
Dr. Samir R. Shah [MBBS, MD - General Medicine, DM - Gastroenterology]
Dr. Sanchika Gupta [MBBS, MD - Dermatology]
Dr. Sandeep Aggarwal [MBBS ( AIIMS), MS ( AIIMS), FACS (USA), FCLS, FAMS, Commonwealth Fellow (UK) Laparoscopic, Bariatric , GI and Robotic Surgeon]
Dr. Sandeep Batra [MBBS, MD - Medicine, DNB - General Medicine, DNB - Medical Oncology]
Dr. Sandeep Bipte [MBBS, MS - General Surgery, Fellowship - Breast Surgery]
Dr. Sandeep Chauhan [MBBS, MS - Orthopaedics, DNB - Orthopedics]
Dr. Sandeep De [MBBS, MD - Radiotherapy]
Dr. Sandeep Goel [MBBS, MD - Radiotherapy, Radiation Oncologist]
Dr. Sandeep Guleria
Dr. Sandeep Harkar [MBBS, MS in Surgery, DNB - Urology/Genito - Urinary Surgery]
Dr. Sandeep K Jha [MBBS, MS - General Surgery, DNB - Surgical Gastroenterology]
Dr. Sandeep Karmarkar [MS ENT]
Dr. Sandeep Kulkarni [MBBS, MD - Internal Medicine, DNB - Gastroenterology]
Dr. Sandeep Kumar Deep [MBBS, MS, FIJR - Joint Replacement]
Dr. Sandeep Mohan
Dr. Sandeep Nayer [MBBS, MD - TB & Chest]
Dr. Sandeep Rai [MBBS, DNB - Internal Medicine, PG Diploma - Diabetes]
Dr. Sandeep Satsangi
Dr. Sandeep Sindhu [MBBS, MS(ENT)]
Dr. Sandeep Singh
Dr. Sandeep Tadas [MBBS, MS (General Surgery), MCh (Cardio Thoracic and Vascular Surgery)]
Dr. Sandeep Vaishya [MBBS, MS - General Surgery, MCh - Neurosurgery]
Dr. Sandhya Vasan
Dr. Sandip Agnihotri [MBBS, DVD, DNB]
Dr. Sandip Ganguly
Dr. Sandip Kumar Bhattacharya
Dr. Sandip Kumar Chandra
Dr. Sandip Mandal
Dr. Sanesh Vijay Tuteja
Dr. Sangeeta Raodeo
Dr. Sangeeta Wagh [MS Ophthalmology]
Dr. Sangeetha Sankaranarayanan
Dr. Sanghamitra Bhattacharya
Dr. Sangita Sharma [MA in Clinical Psychology, MPhil/PhD in Clinical Psychology, AIIMS experience]
Dr. Sanjai V [MBBS, MD - General Medicine, DM - Cardiology]
Dr. Sanjat Chiwane [MBBS, DNB - General Medicine, DM - Cardiology]
Dr. Sanjay Agarwal [MD Medicine, DM Endocrinology]
Dr. SANJAY BHATIA
Dr. Sanjay Bhaumik
Dr. Sanjay Chatterjee
Dr. Sanjay Chaudhary [MBBS, MS (Ophthalmology)]
Dr. Sanjay Chauhan [MBBS, D Ortho, DNB Ortho]
Dr. Sanjay Deshmukh [MS Surgery]
Dr. Sanjay Dhawan
Dr. Sanjay Gandhi [MBBS]
Dr. Sanjay Gogoi [MBBS, MS in General Surgery, MCh - Urology Surgery, DNB - Urology Surgery, MNAMS-Urology]
Dr. Sanjay Helale [MBBS, MS - ENT, Diploma - OtoRhinolaryngology]
Dr. Sanjay Hunugundmath [MBBS, DNB (Radiation Oncology), Fellowship in Stereotactic Radiosurgery, Fellowship in Stereotactic Body Radiotherapy (SRS/SRT/SBRT)]
Dr. Sanjay Jain [MBBS 1977 MAMC, University of]
Dr. Sanjay Joshi [MS General Surgery, MCh Plastic Surgery]
Dr. Sanjay Khanna [MBBS, MD - Medicine, DNB - Gastroenterology]
Dr. Sanjay Kolte [DNB, MNAMS (Gen Sur), FCPS (Gen Sur), MNAMS]
Dr. Sanjay Kumar Gudwani
Dr. Sanjay Kumar [MBBS, MD - General Medicine, DM - Cardiology]
Dr. Sanjay Kumavat
Dr. Sanjay Pandey
Dr. Sanjay Patil [MBBS, MS Orthopaedics]
Dr. Sanjay Raina
Dr. Sanjay S. Nabar
Dr. Sanjay Sachdeva [MBBS, DCH, MS - ENT Surgery]
Dr. Sanjay Sarup [MBBS, MS - Orthopedics, Fellowship, Pediatric Orthopedic Surgeon]
Dr. SANJAY SHAH
Dr. Sanjay Sikka [MBBS, MD(Med), FACG(USA)]
Dr. Sanjay Sobti [MBBS, MD, DNB]
Dr. Sanjay Verma
Dr. Sanjeev Chaudhary [MBBS, MD - General Medicine, DNB - Cardiology]
Dr. Sanjeev Dua
Dr. Sanjeev Gera
Dr. Sanjeev Gulati
Dr. Sanjeev Kapoor
Dr. Sanjeev Kumar Sharma [DM - Clinical Haematology, MBBS, MD - General Medicine]
Dr. Sanjeev Kumar [MBBS, MS - General Surgery (Gold Medalist), MCh - Surgical Oncology, Robotic Surgery]
Dr. Sanjeev Pandey
Dr. Sanjeev Srivastava [MBBS, DNB - General Surgery, DNB - Neurosurgery]
Dr. Sanjeev Y. Vichare
Dr. Sanjeevkumar Ramchandra Kalkekar [MBBS, MD - General Medicine, DNB - Cardiology]
Dr. Sanjiv Badhwar
Dr. Sanjiv Jasuja [MD (Medicine), DNB (Nephrology), MNAMS (Nephrology)]
Dr. Sanjiv KS. Marya
Dr. Sanjna Nayar [MDS , PhD, BDS]
Dr. Sanjoy Biswas
Dr. Sanju Lall [MDS, BDS, FPFA (USA), MFDI, MIDA]
Dr. Sanket Bankar [MBBS, MS General Surgery, MCh Surgical Oncology]
Dr. Sanket Goyal [MBBS (Saurashtra University, Gujarat, 2010), DNB - Paediatrics (National Board of Examination, India, 2014), Fellowship in Neonatology]
Dr. Sankha Subhra Das
Dr. Santanu Acharyya
Dr. Santosh B. Patankar [MBBS, MS - General Surgery]
Dr. Santosh Bhide [MS Ophthalmology]
Dr. Santosh Sontakke [MBBS, DNB General Medicine, DNB Neurology]
Dr. Sapan Vinayak
Dr. Saphalta Baghmar
Dr. Sapna Chopra [MBBS, MD - Transfusion Medicine]
Dr. Sapna Manocha Verma [MBBS, MD]
Dr. Sapna Yadav [MBBS, MD, DNB - Pulmonology]
Dr. Saptarshi Bhattacharya [Dr. DM Endocrinology (AIIMS, New Delhi) MD Medicine (MAMC, New Delhi) MBBS (Kolkata Medical College)]
Dr. Sara Khan [M.Phil Clinical Psychology]
Dr. Sarabpreet Singh [MBBS, MD, IVF & Reproductive Medicine Specialist]
Dr. Saraswati Viswanathan [MBBS, MS in Paediatric Orthopedics]
Dr. Sarat Kumar Sahoo
Dr. Saravanan Periasamy
Dr. Sarbajit Das
Dr. Sarfraz Ahmad [MBBS, PhD, MRCS, FRCS (Urol)]
Dr. Sarika Joshi [MBBS, DMRE, DNB]
Dr. Sarita Bhagwat
Dr. Sarita Gulati [MBBS, MD - Medicine, DM - Cardiology]
Dr. Saroja Balan [MBBS, MRCPCh, FRCP]
Dr. Sarthak Malik [MBBS, MD - Internal Medicine, DM - Gastroenterology]
Dr. Sarthak Moharir [MBBS, MD - Radiation Oncology (Gold Medalist), FCPM]
Dr. Sasmita Vadgaonkar
Dr. Saswati Mukhopadhyay
Dr. Satadru Biswas
Dr. Satheesh Muthuswamy
Dr. Sathish M. S. Vasishta
Dr. Sathiya Rathini SK
Dr. Satinder Kumar Jain
Dr. Satish Khanna [MBBS, MD (MEDICINE)]
Dr. Satish Malegaonkar
Dr. Satish Ratnakar Javali
Dr. Satya Narain Saroha [MBBS, MS - Orthopaedics]
Dr. Satya Prakash Yadav
Dr. Satyakam Baruah [MBBS, MS - General Surgery, MCh - Neurosurgery]
Dr. Satyanarayana Mysore [MBBS, DNB - General Medicine, FRACP (Respiratory Medicine & Sleep Medicine)]
Dr. SATYEN Mehta
Dr. Satyendra Katewa
Dr. Saujatya Chakraborty
Dr. Saurabh Argal [MBBS, DNB - General Medicine, DM - Hepatology]
DR. SAURABH CHANDRA
Dr. Saurabh Gupta
Dr. Saurabh Rawall
Dr. Saurabh Singhal [MS - General Surgery, MBBS]
Dr. Saurabh Varshney
Dr. Saurabh Verma [MBBS, MS - Orthopaedics, FNB - Spine Surgery]
Dr. Saurabh Yatish Bansal [MBBS, MD - General Medicine, DM - Neurology]
Dr. Saveena Gulati Raheja [MBBS, MD, BCCPM- Maulana Azad Medical College]
Dr. Sayan Paul
Dr. Seema Behl
Dr. Seema Dhir [MBBS, DNB - General Medicine]
Dr. Seema Grover [Masters in Physiotherapy]
Dr. Seema Jain [MBBS, DA - Anaesthesia]
Dr. Seema Manuja [MBBS, MD - Obstetrics and Gynecology, Diploma - Advanced Laproscopic Surgery]
Dr. Seema Thakur [MBBS, MS - Obstetrics & Gynaecology, DM - Medical Genetics]
Dr. Seeta Ramamurthy Pal
Dr. Sehriban Diab [MBBS, FRCPC in Rheumatology and Internal Medicine ]
Dr. Selvin S
Dr. Senthil Ganesan
Dr. Senthil Kumar M.S.
Dr. Senthil Kumar TS [MBBS, MD - Anaesthesiology]
Dr. Senthil Kumar TS [MBBS, MD - Anaesthesiology]
Dr. Seral Kannan [MBBS, MS - General Surgery, DNB - Urology]
Dr. Sfurti Mann [MBBS, MD - Internal Medicine]
Dr. Shabber Zaveri [MBBS, MS - General Surgery, MCh - Surgical Oncology]
Dr. Shafiq Ahmed
Dr. Shahid Aboobaker Merchant
Dr. Shahid Mahdi
Dr. Shahin Nooreyezdan [MBBS, MS - General Surgery, MCh - Plastic Surgery]
Dr. Shaikat Gupta
Dr. Shailaja Kale [MD, FRCP (London), FACE, FACP (USA)]
Dr. Shailaja Sabnis
Dr. Shailendra Date [MD (General Medicine)]
Dr. Shailesh Naik [MBBS, DNB General Surgery]
Dr. Shailesh Sable [MBBS, MS - General Surgery, DNB - General Surgery]
Dr. Shaiwal Khandelwal
Dr. Shajathi Begum
Dr. Shakti Bhan Khanna [MBBS, MD]
Dr. Shakti [MBBS, DNB, PGDHIVM, MRCP (UK)]
Dr. Shalab Mohan
Dr. Shaleen Agarwal [MBBS, MS - General Surgery, MCh - Surgical Gastroenterology, G.I. Surgery]
Dr. Shalin Dubey [MBBS, MS - General Surgery, Fellowship - Minimal Access Surgery]
Dr. Shallini Mittal [MBBS, MD - Radiology]
Dr. Shampa Mitra Pahari
Dr. Shankar R
Dr. Shanmugasundaram R [MBBS, MS - General Surgery, DNB - Urology]
Dr. Shantanu Panja
Dr. Shantanu Sharma
Dr. Shantesh Durgesh Kaushik [MBBS, MS - General Surgery, DNB - General Surgery]
Dr. Shanti Talwar
Dr. Shanu Kumar Mittal
Dr. Sharad Bhalekar [MBBS, MS - ENT]
Dr. Sharad Malhotra
Dr. Sharan Choudhri
Dr. Sharan Shivaraj Patil [MBBS, Diploma in Orthopaedics, MS - Orthopaedics, M.Ch - Orthopaedics, FRCS - Trauma & Orthopedic Surgery]
Dr. Sharang Ravindra Wartikar
Dr. Sharat Kumar Das
Dr. Shardul Date [MBBS, FCPS in General Surgery, DNB in Peripheral Vascular Surgery]
Dr. Sharmil Kanna [MD DrNB(CTVS)]
Dr. Sharmila Banwat
Dr. Sharmishtha Patra
Dr. SHASHANK AKERKAR
Dr. Shashank Sharad Kale [MBBS, MS (General Surgery), M.Ch. (Neurosurgery), Reconstructive Spine Fellowship (University of South Florida USA)]
Dr. Shashi Bhushan [MD - Pediatrics, MBBS]
Dr. Shashidhar Shree Niwas [MBBS, DM - Nephrology, MD - Internal Medicine]
Dr. Shashidhar Shree Niwas [MBBS, MD - Internal Medicine, DM - Nephrology]
Dr. Shashidhar TB [MBBS, MS - ENT, Cochlear Implants Surgeon]
Dr. Shashikant Apte [MBBS, MD (General Medicine), FRCPA (Haematology)]
Dr. Shaunak Shah
Dr. Sheetal Mahajani [MBBS, MD - General Medicine, DNB - General Medicine, DM - Gastroenterology]
Dr. Sheetal Mahajani [MD (Medicine), DNB (Medicine), DM (Gastroenterology), DNB (Gastroenterology), Fellowship in Liver Transplantation Medicine (King's College, London, UK)]
Dr. Sheetal Mahey [MBBS, MD, FUP]
Dr. Sheetal Yadav [BDS]
Dr. Shefali Sardana [ DNB -Medical Oncologist, DNB - General Medicine, MBBS]
Dr. Shehla Jamal [MBBS, DGO, DNB, FICOG]
Dr. Sheikh Mohd Murtaza [MBBS, MS, MCh - CTVS]
Dr. Shekhar Shivam
Dr. Shelly Sharma [DNB Radiodiagnosis]
Dr. Shiba Kalyan Biswal [MBBS, MD - Respiratory Medicine]
Dr. Shibal Bhartiya [MBBS, MS - Ophthalmology, Fellowship in Glaucoma & Neuro-ophthalmology]
Dr. Shifa Yadav [MBBS, MD - Dermatology , Venereology & Leprosy, Gold Medalist- MD ]
Dr. Shila Mitra
Dr. Shilpa Gupta [MBBS, DGO]
Dr. Shilpa Jaiswal [MBBS (King George's Medical University), MD - Internal Medicine (King George's Medical University), DM - Neurology]
Dr. Shilpa Naik [MS (General Surgery), FMAS]
Dr. Shilpa S Menon [MBBS, MD - General Medicine, DM - Cardiology]
Dr. Shilpi Budhiraja [MBBS, MS - ENT]
Dr. Shilpy Dolas [MS General Surgery]
Dr. Shirang Sanglikar
Dr. Shirish Yande [MS General Surgery, FRCS]
Dr. Shishir Joshi [MBBS, FCPS (Medicine), MD Internal Medicine (USAIM), Fellowship in Diabetes (Medversity, Liverpool, UK)]
Dr. Shishir Joshi [MBBS, MD - Medicine, Fellowship - College of Physicians and Surgeons]
Dr. Shishir Narayan Shetty [MBBS, MS - General Surgery, MCh - Surgical Oncology]
Dr. Shishir Seth [DM - Clinical Haematology, MBBS, MD - Pathology]
Dr. Shiv Chouksey
Dr. Shiv Kumar Choudhary [MBBS, MS (CTVS), MCh (CTVS - AIIMS, New Delhi)]
Dr. Shiva Priya [MBBS, MD - Obstetrics & Gynaecology]
Dr. Shiva Priya [MBBS, MD - Obstetrics & Gynaecology]
Dr. Shivam Sharma [MBBS, MD - Internal Medicine, Diabetology]
Dr. Shivam Shingla
Dr. Shivam Vatsal Agarwal
Dr. Shivangi Sharma
Dr. Shivani Agarwal [BDS, MDS, MBA]
Dr. Shivanshu Raj Goyal [MBBS, MD - Pulmonary Medicine, DNB - Respiratory Diseases]
Dr. Shiwangi Sharma [MBBS, MD - Microbiology, IDCC]
Dr. Shobana Priya S [MBBS, MD - Obstetrics & Gynaecology]
Dr. Shobha Manish Itolikar
Dr. Shobhit Gupta
Dr. Shoeab Ahmed K.M
Dr. Shomeshwar Singh [MBBS, MS (ENT), DLO (England), FRCS (Ireland), MD (London), Best Cochlear Implant Surgeon in India]
Dr. Shona Nag [MD Medicine]
Dr. Shraddha Shetty [BDS, MDS Orthodontics]
Dr. Shradha Joshi
Dr. Shrestha Sagar Tanwar
Dr. Shrikant Dalal [DNB Ortho, MCh Ortho, MNAMS, FIMISS]
Dr. Shrikant Mhaskar [MBBS, MS (General Surgery), FICS, FAIS (Uro)]
Dr. Shrikant Mhaskar [MS - General Surgery, DNB - Urology/Genito-Urinary Surgery]
Dr. Shrikant Vishwnath [MS Ortho, Fellowship Spine Surgery]
Dr. Shriniwas Kulkarni [MBBS, MD - General Medicine, DM - Medical Oncology]
Dr. SHRIRANG DABHADE
Dr. Shrirang Ranade [MBBS, MS (General Surgery), MCh (CVTS)]
Dr. Shrishu Kamath R
Dr. Shriyansh Jain [MBBS, MD - Medicine]
Dr. Shruti Kharbanda [MD, MBBS]
Dr. Shruti Kohli
Dr. Shubha Bhalla [MBBS, DNB - General Medicine]
Dr. Shubham Jain [MBBS, MS - General Surgery, MCh - Surgical Oncology]
Dr. Shubham Prasad
Dr. Shubhi Tyagi [MS - ENT Surgery, MBBS]
Dr. Shubhra Shri Gupta [MBBS, MD - Pediatrics, Fellowship - Neonatology]
Dr. Shubhranshu S Mohanty
Dr. Shweta Arora [MBBS, DNB - Respiratory Diseases, European Diploma - Adult Respiratory Medicine]
Dr. Shweta Bansal [MBBS, MD - Pulmonary Medicine, DNB - Respiratory Diseases, DM - Pulmonary Medicine & Critical Care Medicine]
Dr. Shweta K. Sharma [MBBS, MS, MCh - Paediatric Surgery]
Dr. Shweta Mehta [MBBS, DGO, FIMS Cosmetic Gynaecology]
Dr. Shweta Mutha [DNB Medicine]
Dr. Shyam Karia [MBBS, MD Anesthesiology, ID]
Dr. Shyam Kishore Shrivastava [MBBS, MD - Radiation Oncology, DNB - Radiation Oncology]
Dr. Shyam Sundar S [MBBS, MS - General Surgery, M.Ch Neurosurgery]
Dr. Shyam Sunder Mahansaria [Liver Transplant & Gastrointestinal Surgeon]
Dr. Shyam Sunder Rendedla [MBBS, DNB - Orthopaedics, Fellowship - Minimal Invasive Spinal Surgery]
Dr. Shyamnath Krishna Pandian K [MBBS, DNB - General Surgery, MCh - Plastic Surgery]
Dr. Sibabrata Banerjee
Dr. Siddharth D
Dr. Siddharth Yadav [MBBS, MS - Orthopaedics and Traumatology, FASIF]
Dr. Siddharth Yande [MBBS, MS (ENT), DNB (Otorhinolaryngology)]
Dr. Siddhartha Gogia [MBBS, MD - Paediatrics, Fellowship in Neonatology]
Dr. Sidharth Sahni
Dr. Simran Sawhney
Dr. Sisir Das
Dr. Sisir Paul
Dr. SITARAM PRASAD
Dr. Siva Prasad Naidu
Dr. Siva Shanmuganathan V
Dr. Sivakami Gopinath [MBBS, MS - Ophthalmology]
Dr. Sivakami Gopinath [MBBS, MS - Ophthalmology]
Dr. SK Jain [MBBS, MS - General Surgery, MCh - Cardiothoracic and Vascular Surgery]
Dr. SK Rajan [MBBS, MS - General Surgery, MCh - Neuro Surgery, Fellow in Spine Surgery, Fellow in Spine Surgery]
Dr. Smita Malhotra [MBBS, DNB (Pediatrics), MRCPCH(UK) IAP Fellowship-Pediatric Gastroenterology and Hepatology]
Dr. Smita Mishra [MBBS, MD - Pediatrics]
Dr. Smita Vats [MBBS, DNB - Obstetrics and Gynecology, Diploma - Advanced Endoscopic Gynae Surgeries]
Dr. Smitha Jain [MBBS, MD - Anaesthesiology]
Dr. Smitha Jain [MBBS, MD - Anaesthesiology]
Dr. Smitha Rukmani [MBBS, MD - Obstetrics & Gynaecology]
Dr. SMRITI NASWA
Dr. Smruti Hindaria [MBBS, MS General Surgery, MCh CVTS]
Dr. Smruti Rajan Mohanty [MBBS, MS - General Surgery, MCh - Cardio Thoracic and Vascular Surgery]
Dr. Sneha A. Deshpande [MBBS, MS - Obstetrics & Gynaecology, DGO, DNB - Obstetrics & Gynaecology]
Dr. Snehal Jhaveri [MBBS, Diploma in Child Health (DCH), FCPS - Child Health, DNB - Paediatrics]
Dr. Snehal M Kulkarni
Dr. Sogani Shani Kumar [MBBS, MS, MCh]
Dr. Sohail Ahmad [MBBS, DNB - Paediatrics]
Dr. Soham Ghosh
Dr. Sohan Lal Broor [MD (General Medicine -PGI Chandigarh) DM (Gastroenterology -PGI Chandigarh) MNAMS (Gastroenterlogy) Fellowship in Gastroenterology ( Medical College of Wisconsin ,Milwaukee, USA)]
Dr. Sohani Verma [MBBS, MRCOG (UK), FRCOG (UK), FICOG, FIMSA, PGDMLS, PGDHHM, FMAS]
Dr. Somesh Virmani
Dr. Somnath Bhattacharya
Dr. Sonal Asthana
Dr. Sonal Kumta
Dr. Sonali Pandit
Dr. Soumen Kar
Dr. Soumya Bhattacharya
Dr. Soumya Ranjan Acharya
Dr. Soumyan Dey
Dr. Sourabh Shirish Kulkarni
Dr. Sovan Sinha
Dr. Sowjanya Aggarwal [MBBS, MS - Obstetrics & Gynaecology]
Dr. Sowmya. M
Dr. Srabani Ghosh Zoha
Dr. Sreekanta Swamy
Dr. Sridevi Murali-Nanavati
Dr. Srinivas Chakravarti Gummaraju
Dr. Srinivas Juluri
Dr. Srinjoy Saha
Dr. Sruthi Sipra Patibandla
Dr. Subash Chandra Bose P
Dr. Subash Rao [MBBS, Diploma - Child Health, DNB]
Dr. Subashree P [MBBS, MD - Obstetrics & Gynaecology]
Dr. Subbiah Shanmugam
Dr. Subhankar Dey
Dr. Subhaprakash Sanyal
Dr. Subhash Chandra
Dr. Subhash Gupta
Dr. Subhash Hakoo
Dr. Subhash Jangid [MBBS, MS - Orthopaedics, DNB - Orthopedics Surgery]
Dr. Subhash Wangnoo [MBBS, FRCP, DM, MD(Medicine)]
Dr. Subhasish Ghosh
Dr. Subhendu Mohanty [MBBS, DM - Cardiology, MD - General Medicine]
Dr. Subir Gangopadhyay
Dr. Subir Ray
Dr. Subodh Chandra Pande [MBBS, DMRE, MD - Radiotherapy]
Dr. Subrat Kumar Acharya
Dr. Subrata Dey
Dr. Subrata Saha
Dr. Suchanda Goswami
Dr. Suchit Majumdar
Dr. Suchita Pant [MBBS, MD - Respiratory Medicine, European Diploma - Adult Respiratory Medicine]
Dr. Sudarsan De [MBBS, MD - Radiotherapy]
Dr. Suddhasatwya Chatterjee
Dr. Sudeep Khanna [MBBS, MD, DM]
Dr. Sudeep Sarkar
Dr. Sudha Kansal [MD, IDCC]
Dr. Sudheer Ambekar [MBBS, MS, MCh - Neurosurgery]
Dr. Sudheer Kumar Tyagi
Dr. Sudheer Pargewar
Dr. Sudhir Bhatia
Dr. Sudhir Dubey
Dr. Sudhir Jadhav [MBBS, MS - General Surgery]
Dr. Sudhir S Pai
Dr. Sudip Roy
Dr. Sudip Sengupta
Dr. Sudipta Kumar Maitra
Dr. Sufla Saxena
Dr. Sugandha Arya [MBBS, MS, MCh - Surgical Oncology]
Dr. Suguna L [General Physician & Diabetologist]
Dr. Suhail Naseem Bukhari
Dr. Suhail Naseem Bukhari [MBBS, DNB - General Surgery]
Dr. Suhas Hardas [MBBS, MD - General Medicine, DM - Cardiology]
Dr. Suhas Hardas [MD (Cardiology)]
Dr. Suhas Vaze [BDS, MDS (Oral & Maxillofacial Surgery)]
Dr. Suhas Wagle [MBBS, MD (Medicine), Fellowship in Gastroenterology]
Dr. Suhas Wagle [MBBS, MD - General Medicine, DM - Gastroenterology]
Dr. Sujai Hegde [MS Surgery]
Dr. Sujata Agarwal
Dr. Sujatha G [MBBS, DGO, MS - Obstetrics & Gynaecology]
Dr. Sujit Chowdhary [MBBS, (AFMC), MS MCh (PGI) FRCS, FACS, MCh Eur Board]
Dr. Sujit Kumar Chowdhary
Dr. Sujoy Kumar Bhattacharjee [MBBS, MS - Orthopaedics, Robotic Orthopaedics Surgery]
Dr. Sukrit Bose
Dr. Sukriti Bhalla
Dr. Sukriti Gupta [MBBS, MD - Pediatric Medicine, Fellowship - Bone Marrow Transplantation]
Dr. Sulakshhana Jagtap [MBBS, MD - Obstetrics & Gynaecology, Diploma - Obstetrics & Gynaecology]
Dr. Suman Bhandari
Dr. Sumana Premkumar
Dr. Sumant Gupta
Dr. Sumanto Mukhopadhyay
Dr. Sumara Maqbool [MBBS, DNB Respiratory, critical care and sleep medicine, DrNB superspeciality Critical care, IDCCM, IFCCM, EDIC]
Dr. Sumedh Magar [MS Orthopaedics]
Dr. Sumeet Agrawal [MD - Medicine, DM - Clinical Immunology, MBBS]
Dr. Sumeet Arora [MBBS, MD Pediatrics (USA), ABP Certified in Pediatric Enodcrinology (USA)]
Dr. Sumeet Sethi
Dr. Sumit Aggarwal
Dr. Sumit Bansal
Dr. Sumit Goyal [MBBS, DNB - Neurosurgery]
Dr. Sumit Gulati
Dr. Sumit Khetarpal
Dr. Sumit Kumar Bose [MD, DIS, FDS (FRANCE), DV DTMSH (UK)]
Dr. Sumit Kumar [MBBS, DNB - Emergency Medicine]
Dr. Sumit Monga
Dr. Sumit Narang [MBBS, MS - General Surgery, MCh - Cardio Thoracic and Vascular Surgery]
Dr. Sumit Paria
Dr. Sumit Sharma [MBBS, MS - General Surgery, MCh - Urology]
Dr. Sumit Singh [MBBS, MD - Medicine, DM - Neurology]
Dr. Sunanda Anand
Dr. Sundeep Kumar Upadhyaya [MBBS, MD - Medicine, DM - Clinical Immunology, Rheumatologist]
Dr. Suneel Chakravarty
Dr. Sunil Almale [MBBS, DNB, DMRD, FRCR (UK)]
Dr. Sunil Bandishti [MD General Medicine, DNB Neurology, DM Neurology]
Dr. Sunil Choudhary
Dr. Sunil G Kini
Dr. Sunil Kaul [MBBS, MS, FELLOW in general minimal access surgery]
Dr. Sunil Kumar Chaudhary
Dr. Sunil Kumar Choudhary
Dr. Sunil Kumar Gupta
Dr. Sunil Kumar Yadav [MBBS, DNB, FIJR - Orthopaedics]
Dr. Sunil Kutty [MBBS, MS - General Surgery, MCh - Neurosurgery]
Dr. Sunil Modi [MBBS (Hons.),MD (Medicine),DM (Cardiology)]
Dr. Sunil Prakash
Dr. Sunil Sanghi [MBBS, MD - Dermatology (AFMC Gold Medalist)]
Dr. Sunil Sathe [MD (Medicine), DNB (Cardiology), DM (Cardiology)]
Dr. Sunil Shahane
Dr. Sunit Mediratta [MBBS, MS, MCh, DNB MNAMS]
Dr. Sunita Gulati [MBBS, MD - Obstetrics & Gynaecology]
Dr. Sunita Tandulwadkar [MBBS, MD Obs & Gynae, DM Reproductive Medicine, FICS, FICOG]
Dr. Sunny Garg
Dr. Sunraj Bangera
Dr. Suparna Ghoshray
Dr. Suparna Telang [MD Psychiatry]
Dr. Supratim Bhattacharyya
Dr. Supreet Kumar
Dr. Supriya Puranik [MBBS, DGO, DNB - Obstetrics & Gynecology]
Dr. Supriyo Ghatak
Dr. Suraj Bhagat Gastroenterology and Hepatology Amrita Hospitals in Faridabad
Dr. Suraj Munjal [MBBS, MS - Ophthalmology]
Dr. Surajit Gorai
Dr. Surakshith T K
Dr. Surender Kumar Dabas [MBBS, MS - General Surgery, DNB - Surgical Oncology , Robotic Cancer Surgery]
Dr. Surendra Pratap Singh
Dr. Surendran R
Dr. Suresh Advani
Dr. Suresh Kalyansundar [MBBS, MS - General Surgery, FIAGES]
Dr. Suresh Kr Rawat [MBBS, MS, MCH, DNB]
Dr. Suresh Kumar Chhabra [MBBS, MS - General Surgery, FMAS]
Dr. Suresh Kumar Rawat
Dr. Suresh Palanisamy Shanmugam
Dr. Suresh Raghavaiah [MBBS, MS - General Surgery, Fellowship - Minimally Invasive Hepatobiliary Surgery]
Dr. Suresh Ramasubban
Dr. Suresh Rao [MBBS, MS General Surgery, MCh Cardio Vascular and Thoracic Surgery]
Dr. Suresh S. Sawardekar
Dr. Suresh Shetty
Dr. Suresh Singh Naruka
Dr. Suresh V. Joshi [MBBS, MS - General Surgery, MS - Cardiothoracic Surgery]
Dr. SURESH VIJAN
Dr. Suresh Vitthalrao Ban [MBBS, DMRD, DNB - Radiology]
Dr. Sureshkumar Bhagat
Dr. Surpreet Kaur Sandhu [MBBS, MS]
Dr. Suruchi Desai
Dr. SURYAPRAKASH BHANDARI
Dr. Suryashree Pandey
Dr. Sushan Mukhopadhyay
Dr. Sushant C Patil
Dr. Sushant Chhabra [MBBS, Master - Emergency Medicine]
Dr. Sushant Srivastava [MBBS, MS - General Surgery, MCh - Cardio Thoracic and Vascular Surgery]
Dr. Sushil Azad
Dr. Sushil Kumar
Dr. Sushil Singla [MBBS, MD - Paediatrics]
Dr. Sushma Prasad Sinha [MBBS, MD, MRCOG(UK), FRCOG(UK), FICOG<FIMSA]
Dr. Sushrut Badve [MBBS, MS (Orthopaedics), DNB (Orthopaedics)]
Dr. Susobhan Mondal
Dr. Suvadip Chakrabarti
Dr. Suvro Banerjee
Dr. Suvro Ganguly
Dr. Suyog Doshi [MBBS, MD - Neurology]
Dr. Suyog Doshi [MD (Internal Medicine), DM (Neurology)]
Dr. Swagat Dash [MBBS (Gold Medal), DNB - Nuclear Medicine, FEBNM]
Dr. Swagata Sarkar [MBBS, MS - Ophthalmology, FMRF]
Dr. Swanand Gholap [MBBS, DORL, FCPS (ORL), MNES (Budapest, Hungary), IVRC (The American Institute of Balance, Florida)]
Dr. Swapan Kumar De
Dr. Swapna Athavale [MBBS, DNB (General Surgery), MCh (Plastic Surgery)]
Dr. Swapna Saha [MBBS, MS - General Surgery]
Dr. Swapnadeep Roy [Mbbs, MS, MCh CTVS]
Dr. Swapneel Karne [MS General Surgery, DNB]
Dr. Swapnil S Patil
Dr. Swapnil Sharma
Dr. Swapnil Shikha [MBBS, MD - Internal Medicine]
Dr. Swapnil Yewalkar [MBBS, MD, DNB (Radiology)]
Dr. Swarup Pal [MBBS, MS - General Surgery , MCh - Cardio Thoracic and Vascular Surgery]
Dr. Swarup Verma
Dr. Swarupa Bansode [MBBS, MD, DNB, DrNB - Neurology]
Dr. Swarupa Bansode [MBBS, MD, DrNB - Neurology]
Dr. Swati Garekar
Dr. Swati Mahajan
Dr. Swati Mohan [MBBS, MD - Dermatology, Venereology & Leprosy]
Dr. Swati Singh
Dr. Swati Suradkar [DNB Surgery]
Dr. Sweta Budyal
Dr. Syamal Kumar Sarkar
Dr. Syamasis Bandyopadhyay
Dr. Syed Afroze Hussain
Dr. Syed Arif Hosain [MBBS, MD Paediatric Neurology, HSSC]
Dr. Syed Harris J
Dr. Syed Ibrahim T
Dr. Syed Uzair [MBBS , M.D. (Internal Medicine), DNB Gastroenterology]
Dr. T S Srinath
Dr. T. Malini
Dr. T. Raja
Dr. T.S. Kler
Dr. Tajamul Syed Malik [Nuclear Medicine]
Dr. Tamal Laha
Dr. Tamashis Mukherjee
Dr. Tanmay Pandya
Dr. Tanmoy Mukhopadhyay
Dr. Tanuj Paul Bhatia
Dr. Tanuja Akre [MBBS, MS - Orthopedics]
Dr. Tanweer Shahid
Dr. Tapan Ghose [MBBS, MD - Medicine, DNB - Cardiology, FNB - Interventional Cardiology]
Dr. Tapan Singh Chauhan [MBBS, MS - General Surgery, MCh - Surgical Oncology]
Dr. Tapas Kumar Kar
Dr. Tapeshwar Sehgal
Dr. Tapish Sahu [MBBS, DNB - General Surgery, DNB - Peripheral Vascular and Endovascular Surgery]
Dr. Tarang K Vora [MBBS, DNB - Neurosurgery]
Dr. Tariq Matin [MBBS, DNB - Neurology]
Dr. Tarlochan Singh Kler
Dr. Tarun Grover
Dr. Tarun Jindal
Dr. Tarun Kapur
Dr. Tarun Lala (PT)
Dr. Tarun Sahni [MBBS, MD, DM]
Dr. Tarun Sharma
Dr. Tarun Singh [MBBS, MS, FIAGES-General Surgery, DrNB, FMAS-Urology]
Dr. Tarun Suri [MBBS, MS (Ortho), FNB (Spine Surgery), DNB]
Dr. Tathagatha Das
Dr. Tej Krishan Thusoo [MBBS, MS - General Surgery]
Dr. Tejal Shetty
Dr. Tejasvi K
Dr. Tejasvi Singh Randhawa [MBBS, MS - General Surgery, MCh - Neurosurgery]
Dr. Tejinder Singh [MBBS, MD - Internal Medicine, DM - Medical Oncology]
Dr. Thanmaya Rai [MS ENT]
Dr. Thichen Kalden Lama
Dr. Thilagavathi Nambi
Dr. Tirathram Kaushik
Dr. TK Jayalakshmi [MBBS, MD - Pulmonary Medicine, Diploma - Environmental, TB & Chest Diseases]
Dr. Trideep Kumar Chaudhary
Dr. Tridibesh Mandal
Dr. Trinanjan Basu
Dr. Trithankar Choudhury
Dr. Trupti Vaishnani [MBBS, DCH - Pediatrics]
Dr. Tushar Aeron
Dr. Tushar Deore [MBBS, D.Ortho, DNB (Orthopaedic Spine Surgery)]
Dr. Tushar Dubey [BDS, MDS - Oral & Maxillofacial Surgery]
Dr. Tushar Madke [MD Medicine, DM Hepatology]
Dr. Tushar Maniar
Dr. Tushar Patil [MBBS, MD (Medicine), DM (Medical Oncology)]
Dr. Tushar Patil [MBBS, MD - Internal Medicine, DM - Medical Oncology]
Dr. Uday Krishna Myneni [MBBS, MS & MCh - Orthopaedics, Robotic hip and knee joint replacement ]
Dr. Uday Phadke [MBBS, MD - General Medicine, DNB - General Medicine, DM - Endocrinology, FACE]
Dr. Uday Phadke [MBBS, MD, DNB, DM (Endocrinology), FACE]
Dr. Uday R Bapat
Dr. Uday Sanglodkar
Dr. Uday Shankar Das
Dr. Uddhavraj Dudhedia
Dr. Ujjawal Kumar [MBBS, MD, DM - Cardiology]
Dr. Ujjawal Kumar [MBBS, MD, DM - Cardiology]
Dr. Uma Mallaiah [MBBS, DO, FRCS]
Dr. Uma Ravishankar [MBBS, DNB, Dip. in Radiation Medicine]
Dr. Umang Shihora [MBBS, MS - Orthopaedics]
Dr. Umesh Gupta
Dr. Upendra Kinjawadekar [MBBS, MD - Pediatrics]
Dr. Urmi Sheth [MBBS, DNB Haematology]
Dr. Urmila Anandh
Dr. Utkarsh Anand [MBBS, MS, MCh - Plastic Surgery]
Dr. Uttam Sidhaye [MD (Anaesthesiology)]
Dr. Uttamkumar Pedhadiya [MBBS, MD - General Medicine]
Dr. V Arun Kumar [M.B.B.S, M.S (PGIMER), M.ch (AIIMS, Delhi)]
Dr. V S V Kumar [MBBS, DNB - Orthopaedics, Fellowship - Reconstructive Orthopaedics]
Dr. V Suresh [BDS, MDS - Oral & Maxillofacial Surgery]
Dr. V Suresh [BDS, MDS - Oral & Maxillofacial Surgery]
Dr. V V Lakshminarayanan
Dr. V. Anand Naik
Dr. V. K jain
Dr. V. M. Durai Mavalavan
Dr. V. P. Bhalla
Dr. V.P Singh
Dr. Vaibhav Choudhary
Dr. Vaibhav Keskar
Dr. Vaibhav Mishra [MBBS, MS - General Surgery, MCh - Cardio Thoracic and Vascular Surgery]
Dr. Vaibhav Sunilkumar Somani [MBBS, DNB - Internal Medicine, DNB - Medical Gastroeneterology]
Dr. Vaibhaw Kumar [HPB & Live Transplant Surgery]
Dr. Vaishali Chouhan [MBBS, MD - Obstetrics & Gynaecology, Diploma - Endoscopy]
Dr. Vaishali Giri [MS Gynaecology, MRCOG]
Dr. Vaishali Lokhande [MBBS, MD - Internal Medicine, DNB - Internal Medicine]
Dr. Vaishali Nayak [DGO, DNB Obstetrics & Gynaecology, FICOG, MNAMS]
Dr. Vaishali Pathak [MBBS, MD (Medicine)]
Dr. Vaishali Ray Srivastava
Dr. Vaishali [MBBS, MD, DM - Cardiology]
Dr. Vandana Gawdi [MBBS, MS - Obstetrics & Gynaecology, DGO]
Dr. Vandana Ghude [MD (Anaesthesiology)]
Dr. Vandana Punjabi
Dr. Vandana Soni
Dr. Vani Kulhalli
Dr. Vanita Arora [M.D, D.N.B. (Cardiology) M.N.A.M.S, FRCP (Edinburgh), FESC, FHRS]
Dr. Vanuli Bajpai [MBBS, MS - Ophthalmology]
Dr. Vardhan Joshi [MBBS, DMRD, DNB, Diploma in MSK USG (UCAM, Spain)]
Dr. Varsha Gorey [MSc - Food & Nutrition, MD]
Dr. Varshit Hathi [MBBS, MD Medicine, DM Cardiology]
Dr. Varun Bansal [DNB (CTVS), FACS (Toronto)]
Dr. Varun Khanna [MBBS, MS - Orthopaedics]
Dr. Varun Mittal [MBBS, MS - General Surgery, MCh - Urology]
Dr. Varun Rehani [MBBS, MD - General Medicine, DNB - Neurology, Advanced Aneurysm Treatment, Epilepsy, Parkinsons Disease]
Dr. Vasundhara Aggarwal
Dr. Vasundhra Sangwan [MBBS, MD - Psychiatry, Diploma in Clinical Psychiatry]
Dr. Vedant Kabra
Dr. Veena Bhat
Dr. Veena Kalra [MBBS, MD - Pediatric Neurologist]
Dr. Veenoo Agarwal [MBBS, MD - Medicine, DM - Medical Oncology]
Dr. Vembu Anand [MBBS (AFMC), MS, DNB (Gen Surg), MCh (Vascular Surgery)]
Dr. Venkatramani V. [MD Paediatrics]
Dr. Vibhore Singhal
Dr. Vibhu Bahl [MBBS, M.S(Ortho)]
Dr. Videesh Sombattina [MBBS, MS, DM - Neonatology]
Dr. Vidhyachandra Gandhi [DNB General Surgery, DNB Surgical Gastroenterology]
Dr. Vidur Jyoti
Dr. Vidya Gupta [MBBS, MRCP, FRCP.(Glasg), FRCPCH(UK)]
Dr. Vidya nair Chaudhry
Dr. Vidyalakshmi [MBBS, MS - General Surgery]
Dr. Vidyalakshmi [MBBS, MS - General Surgery]
Dr. Vignesh Jayabalan
Dr. Vijay Deshmukh
Dr. Vijay Kohli [MBBS, MS - General Surgery, MCh - Cardiovascular and Thoracic Surgery]
Dr. Vijay Kumar
Dr. Vijay Kumar Aggarwal [MBBS, DA - Anaesthesia]
Dr. Vijay Kumar Goyal [MBBS, MD - Radiodiagnosis, EDiR]
Dr. Vijay Sankaran [MBBS, MS - General Surgery, MCh - Neurosurgery]
Dr. Vijay Sharma [MBBS, MS, MCh - Urology & Renal Transplant]
Dr. Vijay Sharma [MBBS, MS, MCh - Urology]
Dr. Vijay Sharnangat
Dr. Vijay Verma [MBBS, MS - ENT]
Dr. Vijay Yewale [MBBS, DCH, MD]
Dr. Vijaya Mohan S [Fellowship in Arthroplasty, DNB in Orthopaedics, D Ortho, MBBS]
Dr. Vijayant Govinda Gupta
Dr. Vijayashree Murthy
Dr. Vijil Rahulan [MBBS, Diplomate in Pulmonary Diseases ]
Dr. Vikas Agarwal
Dr. Vikas Bhardwaj [MBBS, MS - General Surgery, MCh - Neuro Surgery]
Dr. Vikas Choudhary [MBBS, DNB - Radiotherapy]
Dr. Vikas Deshwal
Dr. Vikas Dua [Paediatric Hemato Oncologist and Bone Marrow Transplant specialist]
Dr. Vikas Gupta
Dr. Vikas Gupte
Dr. Vikas Jain
Dr. Vikas Karne [MBBS, MD (Anaesthesia)]
Dr. Vikas Kohli [MBBS, MD - Pediatrics, Diploma - Pediatric Cardiology, American Board Certified Pediatric Cardiologist]
Dr. Vikas Kumar Pandey [MBBS, MD - Radiation Oncology]
Dr. Vikas P Birla [MBBS, DNB, MNAMS, Fellowship in Shoulder and Sports Injury]
Dr. Vikas Sharma
Dr. Vikas Sharma
Dr. Vikas Sharma [MBBS, MD - Internal Medicine (Maulana Azad Medical College, New Delhi), DM - Neurology (PGIMER Chandigarh)]
Dr. Vikas Singhal
Dr. Vikas Yadav [MBBS, MS - General Surgery, Fellowship - Minimal Access Surgery]
Dr. Vikash Jain [MBBS, MD, DNB, DM - Neuro-Radiology Intervention]
Dr. Vikash Kumar Agarwal
Dr. Vikram Barua Kaushik [MBBS, MS - General Surgery, DNB - Urology]
Dr. Vikram Batra Shah
Dr. Vikram Gagneja [MBBS, DNB - Pediatrics, Fellowship - Pediatric Critical Care]
Dr. Vikram Kumar Singh
Dr. Vikram N Raut
Dr. Vikram Paode [MBBS, MS - Orthopaedics]
Dr. Vikram Pratap Singh [MBBS, MS, FRCS]
Dr. Vikram Raut [MBBS, MS, Fellowship - Liver transplant Surgery]
Dr. Vikram Shah is Orthopedics & Joint Replacement Surgeon in Shalby Hospital
Dr. Vikram Sharma
Dr. Vikram Singh Chauhan [MBBS, MS - General Surgery]
Dr. Vikram Singh Yadav [MBBS, DNB - Obstetrics and Gynaecology, Fellowship - Cosmetic Gynae and Sexual Medicine]
Dr. Vikram Tolani [MBBS, MD - Internal Medicine]
Dr. Vikrant Choudhary [MDS, PERIODONTIST]
Dr. Vikrant Kale [MBBS, MRCP, MRCS, FMRF]
Dr. Vinay Goyal
Dr. Vinay Kumar Agarwal
Dr. Vinay Kumar Aggarwal [MBBS, Diploma - Child Health, MD - Pediatric Nephrology]
DR. VINAY KUMAR SHAW
Dr. Vinay Mahendra
Dr. Vinaya Kunjir [MBBS, MD - General Medicine, DM - Rheumatology]
Dr. Vinayak Karmarkar [MD Medicine, DM Cardiology]
Dr. Vindhya Gemaraju
Dr. Vindhya Malasani [MBBS, DNB - Nuclear Medicine]
Dr. Vineet Arora
Dr. Vineet Arya [MBBS, MS - General Surgery , DNB - Peripheral Vascular Surgery]
Dr. Vineet Bhushan Gupta [MBBS, MD, MRCP, DCH]
Dr. Vineet Goel
Dr. Vineet Govinda Gupta [MBBS, MD, DM Medical Oncology (AIIMS - Gold Medalist), ECMO (European Certified Medical Oncologist)
Dr. Vineet Kumar Gupta[ MBBS, MD(Medicine), DNB(Gastroenterology)]
Dr. Vineet Kumar Surana [MBBS, MD - General Medicine, DM - Endocrinology]
Dr. Vineet Narula [MBBS, MS - ENT, DNB - ENT Surgery]
Dr. Vineeta Goel [MBBS, DNB - Radiation Oncology]
Dr. Vineeta Singh [BDS, MDS - Oral & Maxillofacial Surgery]
Dr. Viney Jetley
Dr. Vini Jain [MBBS, DNB - Obstetrics and Gynecology]
Dr. Vinit Shah [MBBS, DNB Internal Medicine, DNB Medical Gastroenterology]
Dr. Vinit Suri [MBBS, MD, DM]
Dr. Vinita Ved Shah
Dr. Vinny Sood
Dr. Vinod Gore [MBBS, MS (Oncology), FAIS, FIAGES]
Dr. Vinod Gore [MBBS, MS - General Surgery, DNB - Surgical Oncology]
Dr. Vinod Raina
Dr. Vinod Sukhija [MBBS, MS, MCh]
Dr. Vinodh Maddireddy
Dr. Viny Kantroo [DNB Resp. Diseases, MRCP, IDCCM, EDARM]
Dr. Vipin Arora [MBBS, MS, FRCS, DU, FRCS]
Dr. Vipin Dehane
Dr. Vipin Gupta [MBBS, DNB - Pulmonology]
Dr. Vipin Gupta [MBBS, DNB - Pulmonology]
Dr. Vipin Khandelwal [MBBS, MD (Pediatrics), Fellowship in Pediatric Hemato-oncology & BMT]
Dr. Vipin Kumar [MBBS, DNB - Emergency Medicine]
Dr. Vipin Maheshwari [MBBS, MS - Orthopaedics]
Dr. Vipul Gupta [MBBS, MD - Radiodiagnosis, Fellowship]
Dr. Vipul Nanda [ MBBS & MS -AIMS ,MCh,MRCS, Plastic and cosmetic Surgeon ]
Dr. Vipul Narain Roy [MBBS, MD, MRCP (UK)]
Dr. Vipul Vijay [MBBS, MS, DNB, Dip SICOT, MNAMS]
Dr. Vipulroy Dayanand Rathod
Dr. Virajrao Kore [MBBS, MD Geriatrics, FICM, DNB]
Dr. Viral Kumar Vasani [MBBS, DNB Neurosurgery]
Dr. Virbhan Balai [MBBS, Diploma - Otorhinolaryngology, MD - General Medicine]
Dr. Vishal A Chafale [MBBS, MD - General Medicine, DM - Neurology]
Dr. Vishal Arora [MBBS, MD - Ophthalmology]
Dr. Vishal Bhasme [MBBS, MS Surgery, MCh Neurosurgery]
Dr. Vishal Chhabra
Dr. Vishal Chhabra
Dr. Vishal Dhir [MBBS, MS - General Surgery, MCh - Cardiothoracic and Vascular Surgery]
Dr. Vishal Garg [MBBS, MD, DM]
Dr. Vishal Gurnani [MBBS, MS - Orthopaedics, Fellowship in Paediatric Orthopaedics]
Dr. Vishal Jain [MBBS, MS - General Surgery, MCh - Neuro Surgery]
Dr. Vishal Jalan
Dr. Vishal K Singh
Dr. Vishal Khandelwal [MBBS, MD - Anaesthesiology, FNB - Critical Care]
Dr. Vishal Khullar
Dr. Vishal Kumar Chorasiya [MBBS, MS, FAIS, FALS (HPB), FICRS, FACS GI Surgery & Liver Transplant]
Dr. Vishal Mangla [MBBS, MS, MCh - Neurosurgery]
Dr. Vishal Naresh Tyagi
Dr. Vishal Rastogi
Dr. Vishal Saxena
Dr. Vishnu Babu G [MBBS, MS - General Surgery, MCh - Plastic Surgery]
Dr. Vishnu Biradar [MD, PDCC Paediatric Gastroenterology]
Dr. Vishnu Hari
Dr. Vishnu Mane [MD Medicine, DM Cardiology]
Dr. Vishvesh Agarwal [MBBS, MS (Ophthalmology), FMRF, FVRF]
Dr. Vishvesh Ashokkumar Agarwal [MS Ophthalmology]
Dr. Vishwanath Jigjinni [MS General Surgery, FRCS]
Dr. Vishwaraj Ratha
Dr. Vivek Barun [MBBS, MD - General Medicine, DM - Neurology]
Dr. Vivek Chaturvedi
Dr. Vivek Garg
Dr. Vivek Goel
Dr. Vivek Gupta [MD, DM, FESC, FEAPCI, FAPSIC,FCSI, FICC, FIC France, FIEIC, FSCAI]
Dr. Vivek Iyer [MBBS, MD - General Medicine, DM - Neurology]
Dr. Vivek Kumar
Dr. Vivek Mittal
Dr. Vivek Mundada [MBBS, DCH, Fellowship - Paediatric Neurology]
Dr. Vivek Mundale
Dr. Vivek Nangia
Dr. Vivek Prakash Aggarwal
Dr. Vivek Raj
Dr. VIVEK SONI
Dr. Vivek Vaibhav [MBBS, MS - Orthopedics]
Dr. Vivek Vasudeo
Dr. Vivek Venkat
Dr. Vivek Vij
Dr. Vivek Vij
Dr. Viveka Kumar
Dr. Vivekanand Sharma
Dr. Vivekanand Singh
Dr. Vivudh Pratap Singh
Dr. W.V.B.S. Ramalingam
Dr. Wasim Khot
Dr. WG CDR Iyengar
Dr. Yadav Munde [MBBS, DNB (Radiology), FVIR]
Dr. Yaja Jebaying [MBBS, MD PEDIATRICS, FELLOWSHIP PEDIATRIC GASTROENTEROLOGY AND HEPATOLOGY AND LIVER TRANSPLANTATION]
Dr. Yajuvendra Gawai
Dr. Yajvender Pratap Singh Rana / Dr. YPS Rana
Dr. Yakub R. Patel [MBBS, MS, FMAS, FIAGES, DIPMAS, MMAS]
Dr. Yash Mathur
Dr. Yashica Gudesar [MBBS, DNB - Obstetrics & Gynecology]
Dr. Yashwanth Singh Tanwar [MBBS, MS(Ortho), DNB(Ortho)]
Dr. Yatin Mehta
Dr. Yatinder Kharbanda
Dr. Yawar Shoaib Ali
Dr. Yogendra Singh Rajput [MBBS, MD - Internal Medicine, DM - Cardiology]
Dr. Yogesh Batra
Dr. Yogita Parashar [MBBS, MD - Obstetrics and Gynecology]
Dr. Yugal K Mishra [MBBS, MS - Surgery, PhD - Cardiovascular Surgery]
Dr. Yugal Kishore Mishra
Dr. Yugal Varandani [MBBS, DNB - Orthopaedics, Fellowship - Arthroscopy & Sports Medicine]
Dr. Yukta Mohta [BHMS, CEM]
Dr. Z S Meharwal
Dr. Zahir Abbas
Dr. Zubin Dev Sharma
Dr.(Brig.) Satish Chandra Mishra [MBBS, MD - General Medicine, DM - Cardiology]
Dr.(Lt Col) Leena N Sreedhar
Dr.(Prof.) Anil Arora
Dr.Arun Kumar Donakonda
Dr.Arvind M Das [MBBS, MD - Medicine, DM - Cardiology]
Dr.Ashish Sabharwal
Dr.Deepak Bhangale
Dr.P.Srinivas Rao
Dr.Prashant Chaudhary
Dr.Rajesh Vukkala
Dr.S.K Sogani
Dr.Shreya Krishna
Dr.Shridhara G
Dt. Diksha Dayal [B.Sc, M.Sc - Nutrition & Dietetics, RD]
Dt. Nashra Gufran [B.Sc, M.Sc - Nutrition & Dietetics]
Lt Col Dr. Rajiv Gupta (Retd) [MBBS, MD - Anaesthesiology, Fellowship - Pain Management]
Major Dr. Rengan
Ms. Malvika Karkare [BSc, PG in Diabetes]
Ms. Meena Kumari [B.Sc - Home Science, M.Sc - Food & Nutrition]
Ms. Rhitika Sharma [MSc, BSc - Nutrition & Dietetics]
Ms. Vaishali Girme [MA (Clinical Psychology), MPhil (Clinical Psychology)]
Parul Yadav [MSc in Clinical Nutrition / Dietetics]
Prof (Dr) Deepak Sreevastava [MBBS, MD - Anaesthesiology, Fellowship in Pain Management]
Prof. (Dr.) J.M. Hans
Prof. Amar Agarwal
Prof. Dr. Mahipal S Sachdev
Prof. Dr. Mohamed Rela
Surg Commodore (Dr.) Sunil Goyal (Retd)
Surgeon Commander Dr. Karthik Madesh Ratnavelu
Vijay Bagul
Reports Attachment